PvP Biologics is on a mission to develop a highly-effective therapeutic to reduce the burden of living with celiac disease. They are advancing an oral enzyme — TAK-062 — designed to break down gluten in the stomach. This exciting research, which began as an iGEM project in 2011, was matured at the Institute for Protein Design before being spun-out into a company in 2016.
Image by Vikram Mulligan
Today, PvP Biologics has announced it has been acquired by Takeda Pharmaceuticals following results from a Phase 1 study.
“Many people living with celiac disease manage their symptoms by following a gluten-free diet, but there is no treatment for those who continue to experience severe symptoms,” said Asit Parikh M.D., Ph.D., Head, Gastroenterology Therapeutic Area Unit at Takeda. “PvP Biologics’ work demonstrated that TAK-062 is a highly targeted therapy that could change the standard of care in celiac disease. We are now applying our deep expertise in gastrointestinal diseases to advance the clinical study of TAK-062 and TAK-101, two programs with different modalities that have both demonstrated clinical proof of mechanism.”
Takeda exercised its option to acquire PvP Biologics for a pre-negotiated upfront payment as well as development and regulatory milestones totaling up to $330 million. Takeda and PvP Biologics previously entered into a development and option agreement, under which PvP Biologics was responsible for conducting research and development through the Phase 1 proof-of-mechanism study of TAK-062 in exchange for funding by Takeda related to a pre-defined development plan.
We are happy to report that the Rosetta molecular modeling suite was recently used to accurately predict the atomic-scale structure of an important coronavirus protein weeks before it could be measured in the lab. Knowledge gained from studying this viral protein is now being used to guide the design of novel vaccines and antiviral drugs.
On January 30, the World Health Organization declared the ongoing coronavirus outbreak (COVID-19, caused by the virus SARS-CoV-2) a public health emergency of international concern. Scientists around the world are racing to learn more about this deadly virus which has already spread to more than 30 countries.
The best predicted structure of the spike protein (blue) closely matches the structure later solved by Cryo-EM (tan).
Importantly, structural biologists are quickly gaining insights into what the proteins that make up this virus look like and how they function.
One viral protein in particular — the spike protein — allows SARS-CoV-2 to fuse its membrane with those on human cells, leading to infection. Researchers at UT Austin this week used cryo-electron microscopy to create the first 3D atomic-scale map of the SARS-CoV-2 spike protein in its prefusion state. Like other viral spike proteins, this spear-like molecule is thought to take on two distinct conformations: one before it infects cells, and a different, ‘post-fusion’ state after. Other groups are also applying similar techniques in their laboratories to learn even more about this critically important protein.
Coronavirus spike proteins — like the proteins found in your body — ‘fold up’ in order to function.
Robetta, our online Rosetta-based protein structure prediction server that is free to use for academics, was able to accurately predict the results of this folding process. In early February, it calculated 3D atomic-scale models of the SARS-CoV-2 spike protein in its prefusion state that closely match those later discovered in the lab.
With this knowledge in hand, researchers at the Institute for Protein Design are now working to create new proteins to neutralize coronavirus. If successful, these antiviral proteins would stick to the SARS-CoV-2 spike protein and thereby prevent viral particles from infecting healthy cells.
A de novo miniprotein binder (pink) designed to bind to the SARS-CoV-2 spike protein.
These new drug candidates — a type of molecule we call ‘mini-protein binders’ — seek to combine the specificity of antibodies with the high stability and manufacturability of small molecule drugs. Mini-protein binders are custom-designed on the computer to adhere only to specific targets, such as specific grooves on the SARS-CoV-2 spike protein.
In 2017 we first reported our high-throughput mini-protein binder design strategy. Together with colleagues we designed and tested over 22,000 mini-proteins that target influenza and botulinum neurotoxin B, along with over 6,000 control sequences to probe contributions to folding and binding, and identified 2,618 high-affinity binders.
The de novo designed mini-protein binders produced in that study exhibited much greater stability at elevated temperatures and better neutralization in animal models than comparable antibodies and natural protein derivatives. Probably as a result of their small size and very high stability, they also elicited little immune response. The best of the flu-targeting designs provide prophylactic (before infection) and therapeutic (after infection) protection against influenza infection in mouse models with a potency rivaling or surpassing that of antibodies.
Our researchers are now designing on the computer tens of thousands of anti-coronavirus mini-protein binders. In the coming weeks we hope to produce these mini-proteins in the lab and measure their ability to bind to spike protein. Following this, much more laboratory testing would still be needed to evaluate the safety and efficacy of these experimental coronavirus drugs.
Designing coronavirus vaccines
Technology developed in the King Lab at the Institute for Protein Design is also being applied to try to create an effective vaccine against SARS-CoV-2.
This experimental SARS-CoV-2 vaccine was made by fusing multiple copies of the coronavirus spike protein (red) to the outside of a designed protein nanoparticle (orange and gray).
Our colleagues in the Veesler Lab in UW Biochemistry and at the Vaccine Research Center at the National Institutes of Health have fused coronavirus spike proteins to the outside of Rosetta-designed protein nanoparticles to form self-assembling vaccine candidates. Some of these are currently being evaluated in mice. This work builds off our recent efforts to create respiratory vaccines by design.
“We are working with our collaborators at UW, the NIH, and the Bill & Melinda Gates Foundation to help create a safe and effective vaccine for not only SARS-CoV-2 but other coronaviruses as well,” said Neil King, who leads the IPD’s vaccine design efforts.
“This outbreak has illustrated that it’s all hands on deck, and all of us together against the bugs, in the fight against infectious disease. The good news is that the community has developed robust methods for antigen design and display over the last several years that are allowing the rapid generation of vaccine candidates that will likely be highly immunogenic.”
With $4 million in matching funds from the National Institutes of Health, the University of Washington has created a new integrated center to match biomedical discoveries with the resources needed to bring innovative products to the public and improve health.
“The University of Washington and regional partner institutions produce some of the most exciting biomedical discoveries and technologies in the world, but we always find it challenging to support their product development as they move into the early commercialization phases,” said the new center’s executive director Rodney Ho, a professor in the UW School of Pharmacy.
UW’s newly funded Washington Entrepreneurial Research Evaluation and Commercialization Hub (WE-REACH), with an annual budget boosted to $1.4 million by contributions from other partners, is organized to mentor and support biomedical entrepreneurs as well as provide project funding to fuel four to six biomedical startups a year with up to $200,000 each. Those projects will include innovative disease treatments, new drugs, diagnostics, genetic testing and health technologies. Ho said the center will support innovation steps not typically supported by research grants, such as human clinical trials or the development of and access to products.
In addition to the NIH, WE-REACH partners include the Institute of Translational Health Sciences, UW CoMotion, the Institute for Protein Design, the School of Pharmacy, and in collaboration with the UW’s Population Health Initiative Innovators will also receive guidance from multiple academic departments and regional institutions. Those institutions include the Fred Hutchinson Cancer Research Institute, Seattle Children’s and other universities in the five states that make up the WWAMI region.
“We are delighted to welcome WE-REACH as a partner,” said Tong Sun, executive director of the Institute of Translational Health Sciences. “At ITHS we are committed to accelerating the translation of discoveries to the clinic. WE-REACH investigators will be able to leverage ITHS programs and resources, so they can help us in our mission to improve health in our communities. This is a very exciting area of translation that we are happy to support.”
WE-REACH is one of five national commercialization hubs selected for funding by the NIH in 2019.
“The journey of biomedical discoveries to products that improve people’s health is expensive and risky. The process requires strategic investment of know-how as well as financial support from public-private partnerships,” said Ho.
“Spinning life science innovations out of research institutions requires expertise and funding that is hard to source in the academic environment,” adds Fiona Wills, assistant vice president, innovation development at CoMotion, UW’s collaborative innovation hub. “WE REACH builds on the infrastructure CoMotion has developed, including our gap fund and training, to provide critical resources needed to de-risk promising technologies into pre-clinical and clinical development.”
The new center will be located in the South Campus Center on the University of Washington’s Seattle campus and at the Institute of Translational Health Sciences in UW Medicine South Lake Union. It will be staffed by Professor Rodney Ho, Executive Director; Terri Butler, Associate Director of Outreach and Partnerships; Matthew Hartman, Coordinator; Christine Jonsson, Administrator; and new hires in project management and technology management roles.
For information on the new center and how to submit a grant submission, please contact Matthew Hartman at WEREACH@uw.edu or 561-339-0676.
The Institute for Protein Design at the University of Washington held the second symposium aimed at providing strategies to address diversity challenges in science, technology, engineering, and math (STEM). The Strategies for Cultivating Inclusion in STEM (SCI-STEM) symposium featured leading keynote speakers, panel discussions, and interactive breakout sessions. Members of the STEM community at all levels, from undergraduates through senior scientists, deans and heads of departments at the university attended.
James E. West, PhD – National Inventors Hall of Fame
Sharon Razovsky, PhD – Increasing Participation of Students with Disabilities in STEM
Undergraduate researcher Radhika Dalal took home a poster award at this year’s Annual Biomedical Research Conference for Minority Students in Anaheim, California. Her mentors include IPD graduate student Una Natterman and Quinton Dowling.
Icosavax, Inc. today announced its launch with a $51 million Series A financing. The company was founded on computationally designed self-assembling virus-like particle (VLP) technology developed here at the IPD (Cell 2019, Preview).
The proceeds of the financing will be used to advance the company’s first vaccine candidate, IVX-121, for respiratory syncytial virus (RSV) for older adults through Phase 1b clinical studies. Icosavax also announced today its leadership team, board of directors and key scientific advisors.
“Icosavax’s vaccine technology solves the problem of constructing and manufacturing VLPs displaying complex antigens by utilizing computationally designed proteins that separate the folding of individual protein subunits from the assembly of the final macromolecular structure. The individual proteins are expressed and purified using traditional recombinant technologies, and then self-assemble into VLPs when mixed together,” said Icosavax co-founder Neil King, Ph.D.
VLPs are known to induce superior immunological responses compared to traditional soluble antigens, eliciting protective immune responses while reducing the need for strong adjuvants, which in some instances have been associated with side effects.
The company’s RSV vaccine candidate, IVX-121, incorporates a stabilized prefusion F antigen licensed from NIAID/NIH (DS-Cav1; Science 2019). Extensive preclinical studies conducted at IPD and Icosavax suggest that IVX-121 could increase the protective immunogenicity of RSV F compared to the DS-Cav1 antigen alone.
IPD-spinout Neoleukin Therapeutics announced this week a merger with Aquinox Pharmaceuticals, a publicly traded company. The combined company will change its name to Neoleukin Therapeutics, and will continue to advance its Rosetta-designed protein platform for cancer, inflammation, and autoimmune diseases.
Neoleukin was spun out of the IPD Translational Investigator Program in January. As a result of this exciting merger, it will be the first publicly traded company in history with a de novo designed protein as its core technology. The new stock ticker will be NASDAQ:NLTX after the deal closes.
As part of the deal, Neoleukin has also gained access to $65 million in capitalization.
“The merger with Aquinox is transformational for our company,” said Neoleukin CEO Jonathan Drachman, MD. “We believe that cytokine mimetics, or Neoleukins, have the potential to offer enhanced therapeutic effects with fewer toxic side effects.”
Senior leadership at Neoleukin still includes three IPD-trainees: Daniel Silva, PhD as VP, Head of Research; Umut Ulge, MD, PhD as VP, Translational Medicine; and Carl Walkey, PhD as VP, Corporate Development. Aquinox’s former stockholders own approximately 61% of the combined company’s capital stock.
Researchers from the IPD and UCSF recently participated in a Reddit Ask Me Anything about LOCKR, our new de novo protein switch. Reddit users had dozens of fantastic questions — so many, in fact, that the team ran out of time before they could address them all.
“The questions were both insightful and interesting,” says Hana El-Samad, a co-senior author of the LOCKR reports. “I had so much fun answering them!”
Hana was joined by Bobby Langan from the IPD and Andrew Ng from UCSF, both co-first authors of the reports. Some participants asked pointed technical questions about concepts that our scientists are already grappling with. Others drew the lens back to ask about the medical and ethical ramifications of making proteins that can control the behavior of cells. (ICYMI: here’s the paper describing LOCKRs design, and here’s how the team turned it into a circuit for cellular feedback.)
Here is our pick for the top five LOCKR questions from our Reddit AMA:
1. How did you guys originally come up with the idea to design these proteins? Would a treatment using LOCKR still have side effects like drugs do? And you used the example of acute inflammation from a TBI; could these proteins be used for other kinds of inflammation as well, such as the chronic inflammation found in autoimmune diseases? – /u/raucous__raconteuse
The idea for LOCKR grew out of a 2016 paper (you may notice some authorship overlap 🙂 ) where we described how to create really well-behaved helical proteins. We wanted to add function into them, so after a couple whiteboard brainstorming sessions, we decided to try to get one part of the protein to switch in the way we published — and install function in such a modular way. Then, within the IPD and with Hana/Andrew, we developed the functions we’ve published and got it to work in living cells! There’s a lot of work still to do to determine if a cell that uses LOCKR will have any unintended side-effects. Of course, we are attempting to engineer the cells in a way to mitigate that in a predictable way.
TBI is an initial indication, but the field of engineering therapeutic cells — especially using LOCKR — is so new that working on other kinds of inflammation and autoimmune diseases is certainly on the table. What indications would you like to see researchers like us work on? – BL
2. Do you guys know yet when LOCKR could be in commercial use? Even a ballpark guestimation would be interesting. – /u/JustTheBP
There is a lot of work that still needs to be done to use LOCKR in a commercially viable product, and that work is starting! Since the biotech/FDA pipeline is (necessarily) long and rigorous, it’ll be many years before something using LOCKR is ready for use in humans. -BL
3. It sounds like the target for the artificial protein is different protein domains. Is there any risk of off-target binding? Does the “key” protein that allows the activity of the artificial protein need to be endogenous? I imagine there could be a situation where it would be desirable to have the artificial protein activated by a pharmaceutical, is that an area of interest for the research or is the focus more on utilizing existing pathways within the cell? – /u/senojsenoj
Because cells are like burritos where everything is mixed together, there is always a risk for off-target interaction, but part of the beauty of LOCKR is that since these proteins were completely designed in a computer, they will be far less likely to interact with other proteins in the cell compared to other engineered proteins that are directly taken from nature. Currently, the Key that activates the Switch is also a designer protein, but many others are interested in designing proteins that are activated by or interact with endogenous proteins. Designing proteins that can be activated by small molecules is also extremely useful, and many others are working on this! -AN
4. What advice do you have for an undergrad, looking to change the world someday? Have any living trials been conducted yet? Will there be any applications in an orthopedic surgical setting, like with joint replacements, to reduce post-op swelling? What about for chronic joint inflammation? Can this also be used in place of immuno suppressants after an organ transplant? – /u/whiskerbizkits
First piece of advice — keep up your passion for changing the world. Second, pursue studies in science and engineering, and think about engaging actively in research (ask professors what research opportunities are available). As to your questions about applications, we believe that live cell therapies (the ability to take cells out of a patient, engineer them and put them back to be “living medicine”) hold great promise for all the areas you mention. For these cells to be safe, effective and robust, they need to be “smart,” which means they need to be able to detect their local environment and react to it. We need to program them to do so. This is where LOCKR (and other synthetic proteins) and synthetic biology in general can help! And btw, these therapeutic cells could also be programmed to shut themselves off once their job is done, so this is not engineering the genetic code of a human, but rather giving them the equivalent of smarter “pills”! –HES
5. How many other names for the protein did you all consider? Did you have to stretch a bit to land on one as cool as LOCKR, or was that just totally serendipitous? – /u/DrColossusOfRhodes
I knew someone would comment on the name! Scott (another co-first author on this paper) and I went through several iterations over the span of a week — he came up with LOCK then I added the R from pRotein considering other, trendy, names in tech right now (CRISPR, tumblr, flickr, grindr, etc). I get a laugh every time I present the acronym. It’s a little stretched… but it works 🙂 -BL
Who’s who:
BL: Bobby Langan, co-first author, UW
AN: Andrew Ng, co-first author, UCSF
HES: Hana El-Samad, co-senior author, UCSF
Today we report in Nature the design and initial applications of the first completely artificial protein switch that can work inside living cells to modify—or even commandeer—the cell’s complex internal circuitry.
The switch is dubbed LOCKR, short for Latching, Orthogonal Cage/Key pRotein.
“In the same way that integrated circuits enabled the explosion of the computer chip industry, these versatile and dynamic biological switches could soon unlock precise control over the behavior of living cells and, ultimately, our health,” said Hana El-Samad, the Kuo Family Professor of Biochemistry and Biophysics at UCSF and co-senior author of the reports.
LOCKR is made of multiple parts. One chain, called the Cage, sequesters a bioactive peptide. Binding of a second molecule, called the Key, to the Cage causes a change in conformation, exposing the peptide. By swapping out the identify of the caged peptide and by tuning binding affinities, different LOCKR switches can be created for a wide range of signaling outputs.
LOCKR can be used to modify gene expression, redirect cellular traffic, degrade specific proteins, and tightly interface with natural proteins. Together with our colleagues at UCSF, we also built new biological circuits that behave like autonomous sensors. These circuits detect cues from the cell’s internal or external environment and respond by making changes to the cell, just as a thermostat senses ambient temperature and directs a heating or cooling system to shut itself off when a desired temperature is reached.
The lead authors of the reports are Bobby Langan and Scott Boyken of the IPD and Andrew Ng of the UC Berkeley-UCSF Graduate Program in Bioengineering. Both Bobby and Scott have gone on to research positions at Lyell.
“Right now, every cell is responding to its environment,” said Bobby. “Cells receive stimuli, then have to figure out what to do about it. They use natural systems to tune gene expression or degrade proteins, for example.”
Bobby and colleagues set out to create a new way to interface with these cellular systems. They used Rosetta to create and tune LOCKR, testing their tool first in vitro then in vivo.
“LOCKR opens a whole new realm of possibility for programming cells,” said Ng. “We are now limited more by our imagination and creativity rather than the proteins that nature has evolved.”
Today we report in Science the identification of hundreds of previously uncharacterized protein–protein interactions in E. coli and the pathogenic bacterium M. tuberculosis. These include both previously unknown protein complexes and previously uncharacterized components of known complexes.
This research was led by postdoctoral fellow Qian Cong and included former Baker lab graduate student Sergey Ovchinnikov, now a John Harvard Distinguished Science Fellow at Harvard.
Augmented by sequences from over 40,000 bacterial genomes, the team assessed coevolution between 5.4 million pairs of E. coliproteins. After finding orthologs and building paired alignments, they used a local statistical model to identify over 21,000 putative protein–protein interactions. Three-dimensional models for proteins in each pair were generated and docked, leading to 804 pairs with the strongest evidence for coevolution.
When compared to predictions inferred from high-throughput experimental screening methods, this new coevolution-based method for identifying protein–protein interactions outperforms in both precision and recall on multiple benchmarks.
814 additional pairs were added to this high-confidence set by incorporating protein pairs reported to interact in experimental studies or on the same operon.
“Coevolution has been useful for understanding how specific proteins interact, but we can now use it as a tool for discovery,” said lead author Qian Cong. “We are going to apply this tool to more pathogens, and the human genome. Our success will depend on how much work other scientists put into annotating which parts of the genome are genes and which parts are something else.”
Today we report the design of synthetic protein arrays that assemble on the surface of mica, a common and exceptionally smooth crystalline mineral. This work, which was performed in collaboration with the De Yoreo lab at PNNL, provides a foundation for understanding how protein-crystal interactions can be systematically programmed.
Our goal was to engineer artificial proteins to self-assemble on a crystal surface by creating an exact match between the pattern of amino acids in the protein and the atoms of the crystal.
“Biology has an amazing ability to organize matter from the atomic scale all the way up to blue whales,” said co-first author Harley Pyles, a graduate student at the Institute for Protein Design. “Now, using protein design, we can create brand new biomolecules that assemble from atomic- to millimeter-length scales. In this case, mica, a naturally occurring crystal, is acting like a big Lego baseplate on top of which we are assembling new protein architectures.”
Rosetta was used to engineer new proteins with customized patterns of electrical charge on their surfaces — new Lego blocks perfectly matched to the mica baseplate. Different designs formed different patterns when deposited on the mica surface, including crowded wires and highly organized honeycomb-like arrays.
“Even though we designed specific atomic-level interactions, we get these structures, in part, because the proteins are crowded out by the water and are forced to pack together,” said James De Yoreo. “This was unexpected behavior and demonstrates that we need to better understand the role of water in ordering proteins in molecular-scale systems.”
By redesigning parts of the proteins, the team was able to produce honeycomb lattices in which they could digitally tune the diameters of the honeycomb pores by just a few nanometers.
Designing atomically precise filaments and lattices from scratch could unlock entirely novel materials and new strategies for synthesizing semiconductor and metallic nanoparticle circuits for photovoltaic or energy storage applications. Alternatively, the protein honeycombs could be used as extremely precise filters, according to co-first author Shuai Zhang, a postdoctoral researcher at PNNL. “The pores would be small enough to filter viruses out of drinking water or filter particulates out of air,” he said.
Citizen scientists can now use Foldit to successfully design synthetic proteins. The initial results of this unique collaboration appear today in Nature.
Brian Koepnick, a recent PhD graduate in the Baker lab, led a team that worked on Foldit behind the scenes, introducing new features into the game that they believed would help players home in on better folded structures.
Players were provided with a poly-isoleucine backbone in a fully extended conformation (60-100 residues in length). They had seven days to fold the backbone into a compact structure and assign a sequence specifying this new structure.
Foldit players produced many creative folds.
Scores in Foldit are calculated using Rosetta. By competing with one another to reach the highest score, Foldit players arrive at virtual proteins with extremely low energies (a high Foldit score corresponds to low protein energy). But since energy alone is not enough for protein design, the Foldit team made adjustments to the Foldit score function. These included requiring the presence of a hydrophobic core, limiting the placement of glycine and alanine, and other side-chain specific terms.
The team experimentally tested 146 Foldit designs. 56 were found to be stable monomers when expressed in E coli. X-ray crystallography and NMR were used to determine the structure of four Foldit designs, which agreed strongly with their design model.
Every step of the way, the team relied on the work of Foldit players to expose problems with the score function. Foldit players are excellent at exploring new kinds of protein folds. For this reason, Foldit players are incredibly helpful for identifying unanticipated weaknesses in Rosetta, and ultimately can improve our understanding of protein folding.
Now that Foldit players can accurately design high-quality proteins from scratch, we can start to challenge Foldit players with more applied protein design problems. Foldit players can now help to design proteins that can assemble into multi-component structures, or that can bind to biological targets as potent medicines, or that can degrade toxic chemicals.
Because Foldit depends on the cooperation and competition of its player community, our scientific ability grows rapidly with the number of Foldit players. We look forward to expanding the Foldit community and recruiting more creative and curious Foldit players!
Today the Baker lab shares some exciting collaborative results of their efforts to design rigid and tunable receptor dimerizers. The first authors of this report are Kritika Mohan, Stanford, and George Ueda, IPD.
From Science:
Exploring a range of signaling
Cytokines are small proteins that bind to the extracellular domains of transmembrane receptors to activate signaling pathways inside the cell. They often act by dimerizing their receptors, and changes in dimer orientation of the extracellular domains can change the signaling output. Mohan et al. systematically explored this tuning effect by designing a series of dimer ligands for the erythropoietin receptor in which they varied the distance and angle between monomers. The topology affected the strength of activation and differentially affected different pathways, which raises the potential for exploiting such ligands in medicinal chemistry.
Natural proteins often shift their shapes in precise ways in order to function. Achieving similar molecular rearrangements by design, however, has been a long-standing challenge. Today, a team of researchers lead by scientists at the IPD report in Science the rational design of synthetic proteins that move in response to their environment in predictable and tunable ways.
The team, which included researchers from UW, HHMI, LBNL, and OSU, set out to create pH-responsive oligomers, or pROs, that self-assemble into designed configurations at neutral pH and cooperatively disassemble at lower pH.
The project was lead by Scott Boyken, a recent postdoctoral fellow in the Baker lab, who used a three-step procedure to create the dynamic proteins: first, parametric design was used to create helical-bundle backbones which were then fitted with histidine-rich hydrogen-bond networks using the HBNet algorithm. Finally, for each pRO, the remainder of the new protein sequence was assigned using Rosetta.
“Designing new proteins with moving parts has been a long-term goal of my postdoctoral work,” said Boyken. “Because we designed these proteins from scratch, we were able to control the exact number and location of the histidines. This let us tune the proteins to fall apart at different levels of acidity.”
Scott Boyken, PhD
Collaborators in the Wysocki Lab at OSU used native mass spectrometry to determine the amount of acid needed to cause disassembly of the proteins. They confirmed the design hypothesis that having more histidines at interfaces between the proteins would cause the assemblies to collapse more cooperatively.
Researchers in the Lee lab were able to show that these pROs can disrupt artificial membranes in a pH-dependent manner, mirroring the behavior of natural membrane fusion proteins which also contain amphipathic helices.
Follow-up experiments with the Lippincott-Schwartz lab showed that these proteins can also disrupt endosomal membranes in mammalian cells, making pROs an attractive tool for engineering the delivery of biologics into the cytoplasm through endosomal escape.
This week we report in NSMB a combined computational design and experimental selection approach for creating proteins that bind selectively to closely related receptor subtypes. This project was led by Luke Dang, a former Baker lab graduate student, and Yi Miao, a postdoctoral researcher in Christopher Garcia’s lab at Stanford.
Abstract:
To discriminate between closely related members of a protein family that differ at a limited number of spatially distant positions is a challenge for drug discovery. We describe a combined computational design and experimental selection approach for generating binders targeting functional sites with large, shape complementary interfaces to read out subtle sequence differences for subtype-specific antagonism. Repeat proteins are computationally docked against a functionally relevant region of the target protein surface that varies in the different subtypes, and the interface sequences are optimized for affinity and specificity first computationally and then experimentally. We used this approach to generate a series of human Frizzled (Fz) subtype-selective antagonists with extensive shape complementary interaction surfaces considerably larger than those of repeat proteins selected from random libraries. In vivo administration revealed that Wnt-dependent pericentral liver gene expression involves multiple Fz subtypes, while maintenance of the intestinal crypt stem cell compartment involves only a limited subset.
This week we report in JACS a general approach for designing self-assembling 2D protein arrays. This project was led by Zibo Chen, a recent Baker lab graduate student, and featured collaborators from the, DiMaio, De Yoreo and Kollman labs at UW.
Abstract:
Modular self-assembly of biomolecules in two dimensions (2D) is straightforward with DNA but has been difficult to realize with proteins, due to the lack of modular specificity similar to Watson-Crick base pairing. Here we describe a general approach to design 2D arrays using de novo designed pseudosymmetric protein building blocks. A homodimeric helical bundle was reconnected into a monomeric building block, and the surface was redesigned in Rosetta to enable self-assembly into a 2D array in the C 1 2 layer symmetry group. Two out of ten designed arrays assembled to μm scale under negative stain electron microscopy, and displayed the designed lattice geometry with assembly size up to 100 nm under atomic force microscopy. The design of 2D arrays with pseudosymmetric building blocks is an important step toward the design of programmable protein self-assembly via pseudosymmetric patterning of orthogonal binding interfaces.
We’re thrilled to share that four members of our Institute have been nominated for the UW Graduate School’s Postdoc Mentoring Award. Each brings invaluable guidance and advice to their graduate student and undergraduate trainees.
This Year’s Winner:
Gabriella Wolff, Biology
Finalists:
Michael Beyeler, Psychology
David Grossnickle, Biology
Matthew Hart, Pathology Karla-Luise Herpoldt, Biochemistry
Kelly Hines, Medicinal Chemistry Parisa Hosseinzadeh, Biochemistry
Kenneth Matreyek, Genome Sciences
Jillian Pintye, Global Health
Julia Ritterhoff, Anesthesiology and Pain Medicine
Nominees:
Ivana Bussi, Biology
Sam Bryson, Civil and Environmental Engineering
Tanvi Deora, Biology
Gilbert Martinez, Physiology and Biophysics
Irene Rembado, Physiology and Biophysics Anindya Roy, Biochemistry
Jon Rueckemann, Physiology and Biophysics Franziska Seeger, Biochemistry
Guozheng Shao, Material Science and Engineering
Han-Wei Shih, Biology
We’ve been selected to join The Audacious Project, a philanthropic collaborative organized by TED. Read all about our project here.
In short, we’re expanding our institute into a global hub of innovation so that protein design can be applied to help solve some of the world’s most pressing challenges.
Our director David Baker gave an inspiring talk live on the TED stage where he laid out his vision for the project.
Support leveraged via The Audacious Project was made possible through the generosity of Laura and John Arnold, Steve and Genevieve Jurvetson, Chris Larsen and Lyna Lam, Lyda Hill Philanthropies, Miguel McKelvey, the Clara Wu and Joe Tsai Foundation, Rosamund Zander and Hansjörg Wyss for the Wyss Foundation, and several anonymous donors.
About The Audacious Project
The Audacious Project surfaces and funds critical projects with the potential to create global change. By removing barriers associated with funding, The Audacious Project empowers social entrepreneurs to dream boldly and take on the world’s biggest and most urgent challenges. Launched last April and housed at TED (the nonprofit devoted to ideas worth spreading), it operates with support from The Bridgespan Group (which consults with nonprofits and investors to accelerate impact). The Audacious Project brings together some of the most respected organizations and individuals in philanthropy — the Skoll Foundation, Virgin Unite, Dalio Foundation, and more.
Today we report in Cell our first computer-designed nanoparticle vaccine targeting respiratory syncytial virus, the primary cause of pneumonia in young children and the leading cause of infant mortality worldwide after malaria.
Although virtually every child will get infected by RSV before the age of three, an estimated 99 percent of deaths associated with the virus occur in developing countries. Despite substantial effort, there is not yet a safe and effective vaccine for RSV.
An international team co-led by researchers at our Institute has generated a first-of-its-kind vaccine that elicits broadly neutralizing antibodies against RSV in mice and monkeys, paving the way for human clinical trials.
“This is the first of many vaccine candidates we have made using this technology,” said senior author Neil King. By swapping out the proteins along the outside of the nanoparticle, Neil’s team hopes to create additional vaccines for diseases as diverse as HIV, malaria, and cancer.
Vaccines by design
Scientists in the King Lab created the vaccine candidate by fusing DS-Cav1, a stabilized version of the viral glycoprotein RSV F which is responsible for membrane fusion, onto their designed two-component protein nanoparticle platform. This yielded a vaccine that can be tuned to display a variable number of antigens by mixing different versions of the purified parts.
Vaccine researchers Brooke Fiala and Neil King in the lab.
With a computer-generated protein nanoparticle at its core, the new multivalent vaccine candidate — dubbed DS-Cav1–I53-50 — is more stable than the trimeric antigen alone. In laboratory testing, it exhibited no discernible loss in antibody binding performance after being stored at elevated temperatures for two weeks. This may translate into a vaccine that does not require refrigeration, greatly reducing the cost and complexity of global vaccine distribution.
The new nanoparticle vaccine based on DS-Cav1 was also ten times more potent in initial tests than DS-Cav1 alone, suggesting it may translate into a more effective vaccine with more durable protection. DS-Cav1 was developed at the National Institute for Allergy and Infectious Disease Vaccine Research Center at the National Institutes of Health and has been shown to elicit significantly higher neutralizing antibody titers than postfusion F in animals and humans. DS-Cav1 is itself being evaluated in a Phase 1 study by NIH as an RSV vaccine candidate.
The RSV vaccine team was led by researchers at UW and the Institute for Research in Biomedicine in Bellinzona, Switzerland. It also included scientists from the Fred Hutch Cancer Research Center in Seattle, USA; the Karolinska Institute in Stockholm, Sweden; the Vaccine Formulation Institute in Godalming, UK; the European Virus Bioinformatics Center in Jena, Germany; the Vaccine Formulation Laboratory at the University of Lausanne, Switzerland; and the Institute of Microbiology at ETH Zürich, Switzerland. The project was funded in part by the Bill and Melinda Gates Foundation.
Read the full report here: https://www.cell.com/cell/fulltext/S0092-8674(19)30109-6 (PDF)
These compact molecules were designed to stimulate the same receptors as IL-2, a powerful immunotherapeutic drug, while avoiding unwanted off-target receptor interactions. We believe this is just the first of many computer-generated cancer drugs with enhanced specificity and potency.
“People have tried for 30 years to alter IL-2 to make it safer and more effective, but because naturally occurring proteins tend not to be very stable, this has proved to be very hard to do,” said a lead author of the paper, Daniel-Adriano Silva, an IPD biochemist. “Neo-2/15 is very small and very stable. Because we designed it from scratch, we understand all its parts, and we can continue to improve it making it even more stable and active.”
“Neo-2/15 has therapeutic properties that are at least as good as or better than naturally occurring IL-2, but it was computationally designed to be much less toxic,” said another lead author, Umut Ulge, an internal medicine physician and IPD biochemist.
Daniel, Umut, Carl Walky and Alfredo Rubio from the IPD have started a company to help bring this exciting drug to market. We wish them the very best in their new venture!
Readers of Nature News & Views selected an article about our work as their 2018 Reader’s Choice. The article, written by Roberto Chica of the University of Ottawa, details our recent publication on de novo fluorescence-activating proteins and explores the challenges of de novo protein design more generally.
From the article:
“The development and application of this computational method for designing β-barrel proteins that bind small molecules is the first demonstration of the de novo design of both protein fold and function, a milestone in the field. Previous computational designs of ligand-binding proteins relied on building a binding cavity into a protein template found in nature, or one that had previously been created in the laboratory. By contrast, Dou and co-workers have designed a β-barrel protein that has a shape distinct from those found in nature, and constructed a binding pocket that is specifically tailored to a target small molecule.
As noted earlier, the authors’ initial designs needed further optimization to identify proteins that have sufficiently high binding affinities for potential applications. More-accurate predictions of protein structures are needed to eliminate the need for such fine-tuning. One way of achieving this might come from recognizing that proteins are not rigid molecules that adopt a single predominant structure — like all machines, proteins need to move to accomplish their tasks with high efficiency5,6. Indeed, ligand binding is often the trigger that causes a protein receptor to undergo a structural change enabling the transmission of a biological signal7. Computational methods for the rational design of proteins that undergo particular structural changes have recently been developed8. If these could be combined with Dou and colleagues’ approach, it might be possible to access more-complex protein functions than were previously possible, opening the door to the on-demand creation of protein-based molecular machines.”
We thank Roberto and the News & Views readers for their interest in our work.
To close out the year, Baker Lab scientists published a new report describing the creation of proteins that mimic DNA. We believe this breakthrough will aid the creation of bioactive nanomachines.
DNA is a widely used building material at the nanoscale because it is simple and predictable: A pairs with T and C pairs with G. Because of this, DNA strands can be programmed to click together into precise and increasingly complex structures. But DNA has drawbacks. It is not as bioactive as RNA, and not nearly as active as proteins. Bioactive protein assemblies run cells (kinetochores, polymerases, proteasomes, etc). What if designing them was as easy as clicking together DNA?
Using computational design, we created heterodimeric proteins that form double helices with hydrogen-bond mediated specificity. When a pool of these new protein zippers gets melted and then allowed to refold, only the proper pairings form. They are all-against-all orthogonal. With these new tools in hand, we can now begin constructing large protein-based machines that self-assemble in predictable ways.
This project was led by graduate student Zibo Chen and was done in collaboration with the Wysocki Lab at Ohio State University and the Sgourakis Lab at the UC Santa Cruz. The work used support from the SIBYLS program with SAXS and the ALS resources at LBNL, as well as the Argonne Leadership Computing Facility at ANL.
Dr. Ingrid Pultz, an IPD Translational Investigator and Chief Scientific Officer at PvP Biologics, has written a special report for the American Council on Science and Health about how protein design is being used to help fight celiac disease. Pultz describes how an international competition, a video game, and venture capital all aligned to help enable this exciting work.
The basic parts of proteins — helices, strands and loops — can be combined in countless ways. But certain combinations are trickier than others. This week scientists from the IPD, along with collaborators in Brno and Santa Cruz, published the first-ever example of designed non-local beta-strand interactions.
Beta-sheet proteins carry out critical functions in biology, and hence are attractive scaffolds for computational protein design, but the de novo design of all-beta-sheet proteins from first principles has lagged far behind the design of all-alpha or mixed-alpha-beta domains.
Tamuka M. Chidyausiku, a biochemistry graduate student, was one of the project leaders.
Local beta-strand interactions occur when residues near one other hydrogen bond to form compact sheets. To get similar interactions from stretches of residues that are not close in primary sequence, a protein backbone must fold into a complex interwoven shape. The successful design of non-local beta-strand interactions demonstrates a significant advance in our ability to control both fine features (such as precise hydrogen bonding) and global features (such as complex topology) in proteins and opens the door to the design of a broad range of non-local beta-sheet structures.
By studying loops that connect unpaired beta-strands (beta-arches), the team identified a series of structural relationships between loop geometry, side chain directionality and beta-strand length that arise from hydrogen bonding and packing constraints on regular beta-sheet structures. They used these rules to de novo design jellyroll structures with double-stranded beta-helices formed by eight antiparallel β-strands. NMR of a hyperthermostable design closely matched the computational model, demonstrating accurate control over the beta-sheet structure and loop geometry.
In the summer of 1961, Osamu Shimomura drove across the country in a cramped station wagon to scoop jellyfish from the docks of Friday Harbor. He wanted to discover what made them glow.
It took Shimomura and other biochemists more 30 years to find a full answer. By then, recombinant DNA technology allowed researchers to clone and characterize the two proteins responsible: aequorin and GFP. The latter would earn Shimomura his share of the 2008 Nobel Prize.
GFP, a 238-residue beta-barrel with a covalently linked chromophore, transformed how scientists study cells and the molecules in them. As a genetic tag, GFP has illuminated the inner workings of human brain cells, bacteria, fungi and more.
This week, scientists from the IPD report in Nature the design of a completely artificial fluorescent beta-barrel protein.
Comparison of of GFP (left) and the new fluorescent protein (right) a, Surface mesh and ribbon representations. b, Close-up of chromophore binding interactions.
Many natural proteins evolved to bind small molecules. Reengineering such proteins is rarely straightforward, limiting how they can be applied. The new findings demonstrate that proteins unlike any found in nature can be rationally-designed to bind to and act on specific small molecules with high precision and affinity.
The lead authors of the paper are Jiayi Dou, Ph.D and Anastassia A. Vorobieva, Ph.D., then both senior fellows in the Baker lab.
Anastassia Vorobieva with her son Alexandre (left) and Jiayi Dou (right).
To make the fluorescent protein the researchers had to achieve another first: Creating beta-barrels from scratch. The fold was ideal because one end of its cylindrical shape could be used to stabilize the protein while the other could be used to create a cavity that would serve as the binding site for the target molecule, DFHBI. In nature, beta-barrels proteins bind a wide range of small molecules.
Rosetta was used to design the scaffold de novo. To create the cavity, the team used a new docking algorithm called the “Rotamer Interaction Field” or RIF, developed by William Sheffler, Ph.D., a senior research scientist in the Baker lab, that rapidly identifies all potential structures of cavities that fulfill the prerequisites for binding specific molecules.
The designed protein absorbs blue and emits cyan light. It is stable up to 75°C.
“It worked in bacterial, yeast and mammalian cells,” said Dou, “and being half the size of green fluorescent protein should be very useful to researchers.”
“Equally important,” Baker added, “it greatly advances our understanding of the determinants of protein folding and binding beyond what we have learned from describing existing protein structures.”
It was a great year for the Institute for Protein Design and we couldn’t have done all of our amazing work without the support from our donors and contributors! Thank you to everyone who helped us, whether through a donation, collaboration, playing Foldit, or otherwise. We’ve filled the IPD Newsletter with all of the progress we’ve made in 2018, so take a look! In the PDF there are links to articles and publications, but many of them can also be found on either this website, or at www.bakerlab.org. Please continue to watch our growth as we head into 2019!
The funds will support our technological revolution in protein design and enable the development of a universal flu vaccine.
The $11.3 gift is one of the largest made to date by the San Francisco-based philanthropy in support of science. It is also the first to go to UW Medicine.
The gift comes in two parts:
$5.6 million to refine and advance Rosetta, our software platform for protein design.
$5.7 million for our universal flu vaccine design program.
“We’re excited to help move science forward in ways not seen before with proteins, which are essential to life. This grant recognizes that UW Medicine is at the forefront of unlocking the keys to the use of proteins in medical settings,” says Chris Somerville, a Program Officer for Scientific Research at the Open Philanthropy Project. “The universal flu vaccine is a tough nut to crack, but David Baker has shown the ability to pioneer life-changing scientific research. It’s exciting that whether a universal flu vaccine is developed or not, this gift will build techniques and technologies that will advance science and have a huge variety of implications in medicine and industry.”
Proteins are the workhorses of all living creatures, fulfilling the instructions of DNA. Existing proteins are the products of billions of years of evolution and carry out all the important functions in our body—digesting food, building tissue, transporting oxygen through the bloodstream, dividing cells, firing neurons, and powering muscles.
“This gift is speeding up a technological revolution in how we design proteins. Our team can now custom design proteins from scratch, creating entirely novel molecules that can be used for new treatments, new diagnostics and new biomaterials. The Open Philanthropy Project’s generous gift will transform our ability to design proteins from scratch,” said David Baker, the institute’s director as well as professor of biochemistry at the University of Washington School of Medicine and Howard Hughes Medical Institute investigator. Baker is the Henrietta and Aubrey Davis Endowed Professor in Biochemistry.
Computer-based protein design
The gift will accelerate the institute’s efforts to advance protein design on computers with the Rosetta software originally developed in Baker’s lab. Baker said the gift will transform the institute’s ability to design proteins on computers, test them by creating the actual proteins in the lab, and then repeat the process at an enormous scale. “By speeding up this cycle of design, building, and testing, we will be able to systematically improve protein design methods,” Baker said.
The results and new Rosetta software will be shared with the scientific community through the Rosetta Commons. The Rosetta Commons is a collaboration founded by Baker with almost 100 developers from 23 universities and laboratories who regularly contribute to and share the Rosetta source code, currently over 3 million lines.
This project is in collaboration with Frank DiMaio, assistant professor of biochemistry at the University of Washington School of Medicine.
Universal flu vaccine
Current flu vaccines are intended to protect only against currently circulating strains, requiring the vaccines to be reformulated every year as the virus mutates, and are only partially protective. With Open Philanthropy Project support, Baker and his collaborators, Neil King and David Veesler, both assistant professors of biochemistry at the University of Washington School of Medicine, will be leading an effort to design universal flu vaccine candidates that provide durable protection against multiple virus strains, including strains that have the potential to cause pandemic outbreaks. The vaccine candidates will be based on the self-assembling protein nanoparticle technology Baker and King have developed. To ensure that the vaccine candidates are thoroughly and efficiently tested, they will work in close collaboration with the groups of Dr. Barney Graham and Dr. Masaru Kanekiyo at the Vaccine Research Center of the National Institute of Allergy and Infectious Diseases at the National Institutes of Health.
The goal is to design a nanoparticle vaccine that can trigger an effective immune response to many existing flu strains as well as new strains that might appear in the future. Researchers hope such a universal vaccine might need to be administered no more than every five years, ending the need for annual flu vaccinations.
About the Open Philanthropy Project
The Open Philanthropy Project identifies outstanding giving opportunities, makes grants, follows the results, and publishes its findings. Its main funders are Cari Tuna and Dustin Moskovitz, a co-founder of Facebook and Asana.
Update 2018-07-26: The 2018 SCI-STEM Symposium was recently featured in an eLife article.
The Institute for Protein Design at the University of Washington held its first ever symposium aimed at providing strategies to address diversity challenges in science, technology, engineering, and math (STEM). The Strategies for Cultivating Inclusion in STEM (SCI-STEM) symposium featured leading keynote speakers, panel discussions, and interactive breakout sessions. Members of the STEM community at all levels, from undergraduates through senior scientists, deans and heads of departments at the university attended.
As a technical institute dedicated to the pursuit of knowledge and discovery, we know first-hand that innovation in STEM requires bringing in new perspectives to difficult problems. Research groups that create and successfully maintain workplaces where all voices are heard will continue to outperform those that don’t. This website showcases some of the conversations and lectures held at this inaugural SCI-STEM and we hope the extended community at UW and beyond can benefit from the practical tools, data driven ideas and methods proposed towards cultivating a more inclusive workplace. We invite you to keep the conversation going on social media using the hashtags #diversifySTEM .
Dr Hannah Valantine – NIH Addresses the Science of Diversity: Focusing on Institutional Change
Designed Membrane Protein: (Left) Side view showing the designed membrane protein inside the membrane. (Right) Top view of same.
The Abstract reads as follows:
The computational design of transmembrane proteins with more than one membrane-spanning region remains a major challenge. We report the design of transmembrane monomers, homodimers, trimers, and tetramers with 76 to 215 residue subunits containing two to four membrane-spanning regions and up to 860 total residues that adopt the target oligomerization state in detergent solution. The designed proteins localize to the plasma membrane in bacteria and in mammalian cells, and magnetic tweezer unfolding experiments in the membrane indicate that they are very stable. Crystal structures of the designed dimer and tetramer—a rocket-shaped structure with a wide cytoplasmic base that funnels into eight transmembrane helices—are very close to the design models. Our results pave the way for the design of multispan membrane proteins with new functions.
At the end of a historic year for protein design, the Baker Lab was honored to be profiled in the New York Times by science writer Carl Zimmer. He writes about the technology, progress, and promise in the field, including the contributions from our wonderful crowdsource participants.
Graphic: John Hersey / New York Times
On the technology front, Rosetta continues to improve each year thanks to the hard work of the RosettaCommons. Given ongoing advances in DNA synthesis and protein screening technology, we believe there is still so much more we can design and discover.
Progress in the field of protein design was staggering in 2017. Thousands of novel proteins were designed and manufactured with new features, folds, and functions. From immunotherapy, drug delivery, antiviral activity,y and more, this was an awesome year for applied protein design.
The promise of protein design has never been greater. “As we understand more and more of the basic principles, we ought to be able to do far better.”
Today marks another major step forward for peptide based drug discovery. IPD researchers report in Science the computational design of a new world of small cyclic peptides, “Macrocycles”, increasing the number of the known kinds of these molecules by multiple fold. The conceptual art image below “Illuminating the energy landscape” shows the power of computational design to explore and illuminate structured peptides across the vast energy landscape.
Small peptides have the benefits of small molecule drugs, like aspirin, and large antibody therapies, like rituximab, with fewer drawbacks. They are stable like small molecules and potent and selective like antibodies.
Image by Vikram Mulligan. Computational design calculations reveal the peptide macrocycle energy landscape.
Abstract reads as follows.
Mixed-chirality peptide macrocycles such as cyclosporine are among the most potent therapeutics identified to date, but there is currently no way to systematically search the structural space spanned by such compounds. Natural proteins do not provide a useful guide: Peptide macrocycles lack regular secondary structures and hydrophobic cores, and can contain local structures not accessible with L-amino acids. Here, we enumerate the stable structures that can be adopted by macrocyclic peptides composed of L- and D-amino acids by near-exhaustive backbone sampling followed by sequence design and energy landscape calculations. We identify more than 200 designs predicted to fold into single stable structures, many times more than the number of currently available unbound peptide macrocycle structures. Nuclear magnetic resonance structures of 9 of 12 designed 7- to 10-residue macrocycles, and three 11- to 14-residue bicyclic designs, are close to the computational models. Our results provide a nearly complete coverage of the rich space of structures possible for short peptide macrocycles and vastly increase the available starting scaffolds for both rational drug design and library selection methods.
Published today in Nature, IPD researchers describe the first synthetic protein assemblies — dubbed synthetic nucleocapsids — that encapsulate their own genome and evolve in complex environments.
Computationally Designed Synthetic Nucleocapsid, Illustration by Institute for Protein Design & Cognition Studio
Synthetic nucleocapsids are built to resemble viral capsids and could be used in future to deliver therapeutics to specific cells and tissues. These icosahedral protein assemblies are based off of previously reported results from the Institute for Protein Design.
The image above visualizes the de novo creation of synthetic nucleocapsids from computationally designed proteins and their evolution to acquire properties that could be useful for drug delivery and other biomedical applications. The narrative was designed as a futuristic hologram projection realized through spiraling DNA composed of binary zeros and ones. The projection and computational imagery evoke futuristic technology and design, while calling out natural evolution through the DNA spiral “time-scale” motif. The heads-up display iconography showing a blood bag, mouse, and RNase A convey the challenges we used to evolve the synthetic nucleocapsids. The single net impression of this image is engaging + enlightening and shows that we are entering the next epoch of synthetic biology in which biological systems can be designed and created from scratch.
Abstract:
The challenges of evolution in a complex biochemical environment, coupling genotype to phenotype and protecting the genetic material, are solved elegantly in biological systems by the encapsulation of nucleic acids. In the simplest examples, viruses use capsids to surround their genomes. Although these naturally occurring systems have been modified to change their tropism and to display proteins or peptides, billions of years of evolution have favoured efficiency at the expense of modularity, making viral capsids difficult to engineer. Synthetic systems composed of non-viral proteins could provide a ‘blank slate’ to evolve desired properties for drug delivery and other biomedical applications, while avoiding the safety risks and engineering challenges associated with viruses. Here we create synthetic nucleocapsids, which are computationally designed icosahedral protein assemblies with positively charged inner surfaces that can package their own full-length mRNA genomes. We explore the ability of these nucleocapsids to evolve virus-like properties by generating diversified populations using Escherichia coli as an expression host. Several generations of evolution resulted in markedly improved genome packaging (more than 133-fold), stability in blood (from less than 3.7% to 71% of packaged RNA protected after 6 hours of treatment), and in vivo circulation time (from less than 5 minutes to approximately 4.5 hours). The resulting synthetic nucleocapsids package one full-length RNA genome for every 11 icosahedral assemblies, similar to the best recombinant adeno-associated virus vectors. Our results show that there are simple evolutionary paths through which protein assemblies can acquire virus-like genome packaging and protection. Considerable effort has been directed at ‘top-down’ modification of viruses to be safe and effective for drug delivery and vaccine applications; the ability to design synthetic nanomaterials computationally and to optimize them through evolution now enables a complementary ‘bottom-up’ approach with considerable advantages in programmability and control.
Today, scientists from of the Institute for Protein Design will join Foldit gamers from around the world to help design an enzyme that can neutralize aflatoxin — a cancer-causing toxin produced by certain fungi that are found on agricultural crops such as corn, peanuts, cottonseed, and tree nuts. Foldit is a citizen science game version of Rosetta@home, that allows gamers to create new proteins. Aflatoxin puzzles provide a starting enzyme which has the potential to disarm the toxin, and Gamers from around the world will compete to redesign the enzyme so it can neutralize aflatoxin.
Food safety is a long-standing interest at the Institute for Protein Design. Our scientists have designed a potent KumaMax enzyme for breaking down gluten, and have launched PvP Biologics to develop it as a therapeutic for treating celiac disease.
David Baker, director of the UW Institute of Protein Design whose lab has been developing FoldIt along with the UW Center for Game Sciences and Seth Cooper at Northeastern, said: “It has been fascinating to work with FoldIt players over the years and see how they have been able to come up with innovative solutions to challenging problems. I look forward to seeing the solutions FoldIt players come up with to the important aflatoxin neutralization problem!”
Foldit is a competitive online puzzle game about protein folding. It is a crowd-sourcing computer game that allows anyone in the world with a computer and imagination – but not necessarily any scientific training – to determine how amino acids are folded together to create proteins, the workhorses of our bodies.
Inspired by citizen scientists who had a desire to fold proteins on their own, the Foldit game was first released in May 2008, the result of a UW collaboration between David Baker (Director of the Institute for Protein Design), Zoran Popović (Professor of Computer Science), and Seth Cooper (now at Northeastern University). The first Foldit players had previously volunteered their home computers for the Rosetta@home project to support large scale protein folding calculations for the Baker lab, but these first players wished to do with their minds what their Rosetta@home computer was attempting as viewed through a screen saver. Since the launch of the Foldit game, players have had a number of notable successes.
Mark your calendars! September 27, 2017 is the day the doors opened to whole new world of targeted therapeutics. The Baker lab and numerous talented collaborators published in Nature that it is now possible to conduct “Massively parallel de novo protein design for targeted therapeutics”. Three factors make this possible: Rosetta molecular modeling algorithms for computational protein design, economical computing power, and inexpensive gene write – read technology. Designer therapeutic mini-proteins have arrived!
Artist impression of designed mini-protein binders targeting Influenza hemagglutinin to effectively bind and neutralize the virus.
The group designed and tested 22,660 mini-proteins of 37–43 residues that target influenza haemagglutinin and botulinum neurotoxin B, along with 6,286 control sequences to probe contributions to folding and binding, and identified 2,618 high-affinity mini-binders. Comparison of the binding and non-binding design sets, which are two orders of magnitude larger than any previously investigated, enabled the evaluation and improvement of the computational model. Biophysical characterization of a subset of the binder designs showed that they are extremely stable and, unlike antibodies, do not lose activity after exposure to high temperatures. The designs elicit little or no immune response and provide potent prophylactic and therapeutic protection against influenza, even after extensive repeated dosing. This design capability opens the door to a whole new future of genetically encoded, tailor made protein therapeutics. Its a bright new day.
The news of this breakthrough has been highlighted by GEN, CEN, Science Daily and others.
It was a great year for the Institute for Protein Design and we couldn’t have done all of our amazing work without the support from our donors and contributors! Thank you to everyone who helped us, whether through a donation, collaboration, playing Foldit, or otherwise. We’ve filled the IPD Newsletter with all of the progress we’ve made in 2017, so take a look! In the PDF there are links to articles and publications, but many of them can also be found on either this website, or at www.bakerlab.org. Please continue to watch our growth as we head into 2018!
Today, the first IPD spin out company Cyrus Biotechnology announced the closing of an $8M total Series A financing. The investment was led by Trinity Ventures, with participation from OrbiMed Advisors, SpringRock Ventures, W Fund, WRF Capital (a major supporter of the IPD), and individual investors. Congratulations Cyrus team!
Cyrus is commercializing Cyrus Bench® an innovative user friendly software as a service (SaaS) cloud computing solution for distribution of the powerful “Rosetta” protein structure prediction and design algorithms.
The company’s name was inspired by Cyrus Levinthal’s famous paradox, that most small proteins fold spontaneously on short time scales of less than a millisecond, despite there being are a very large number of degrees of freedom in an unfolded protein chain of amino acids, leading to an astronomical number of possible conformations that may need to be sampled before folding into a low energy conformation. The Rosetta suite of algorithms that originated over 17 years ago at the UW, now with a team of over 100+ programmers contributing to the Rosetta Commons, in many cases solves Levinthal’s paradox.
In the last 9 months IPD / Baker lab related Seattle area spin out companies have raised in excess of $55 million to commercialize innovations in computational protein modeling and design !
Today, Baker lab spin out company Arzeda announced that it had raised $12 million in a Series A round of funding led by OS Fund and including Bioeconomy Capital and Sustainable Conversion Ventures, as well as a follow-on investmentfrom Arzeda’s seed investor, WRF Capital (a major supporter of the IPD). The new funding will enable “Technology scale-up that will unlock production of proteins that create sustainable stain-resistant paint, stronger Plexiglas, next-generation sweeteners, and purpose-built molecules that don’t yet exist”
NOTE: Nov. 28, 2017, Arzeda expanded the Series A to $15.2 million, see news.
The Matrix movie (1999) depicts a future in which the reality perceived by most humans is actually a computer simulated reality called “the Matrix”. Published today in Science, the Baker lab and collaborators report on a new kind of Matrix – a new reality for large scale computational protein design which can achieve massive data driven improvements in our ability to design highly stable, small proteins from scratch.
Illustration by Gabe Rocklin
Following the White Rabbit, Postdoctoral fellow Dr. Gabe Rocklin led a group of scientists to design and test over 15,000 new mini-proteins (which do not exist in nature) to see whether they form stable folded structures. Even major protein design studies in the past few years have generally examined only 50 to 100 designs. Synthetic DNA technology and high throughput screening permitted the group to conduct large-scale testing of structural stability of multitudes of computationally designed proteins. In turn, this allows them to perform a “Global analysis of protein folding using massively parallel design, synthesis and testing“.
Through iterative improvements in the design process, the group arrived at 2,788 stable mini-protein structures, which is at least 50-fold more proteins than have ever been characterized from natural sources for similar sized proteins. Their small size and stability may be advantageous for treating diseases when the drug needs to avoid the immune system and reach the inside of a cell.
The publicationAbstractis a step into the Matrix as Morpheus explains,
Proteins fold into unique native structures stabilized by thousands of weak interactions that collectively overcome the entropic cost of folding. Though these forces are “encoded” in the thousands of known protein structures, “decoding” them is challenging due to the complexity of natural proteins that have evolved for function, not stability. Here we combine computational protein design, next-generation gene synthesis, and a high-throughput protease susceptibility assay to measure folding and stability for over 15,000 de novo designed miniproteins, 1,000 natural proteins, 10,000 point-mutants, and 30,000 negative control sequences, identifying over 2,500 new stable designed proteins in four basic folds. This scale — three orders of magnitude greater than that of previous studies of design or folding—enabled us to systematically examine how sequence determines folding and stability in uncharted protein space. Iteration between design and experiment increased the design success rate from 6% to 47%, produced stable proteins unlike those found in nature for topologies where design was initially unsuccessful, and revealed subtle contributions to stability as designs became increasingly optimized. Our approach achieves the long-standing goal of a tight feedback cycle between computation and experiment, and promises to transform computational protein design into a data-driven science.
Today, a multidisciplinary team of researchers at the University of Washington, Fred Hutch, and The Scripps Research Institute published in Nature Biotechnology the computational design of a trimeric influenza-neutralizing protein that binds extremely tightly to the H3 hemagglutinin of 1968 Hong Kong pandemic influenza virus (A/Hong Kong/X31/1968). It also cross-reacts with human relevant H1, H2 and H3 influenza strains.
Figure 1. Design Process for Flu-Glue, a Potent Computationally Deigned Anti-Flu Protein.
The research has been recognized by opinion leaders and media outlets as a major step in the fight against the flu. See articles in Science, the Conversation, and Scientific American, C&EN News.
Crafted in the Baker lab at the Institute for Protein Design, the protein affectionately known “Flu-Glue” was shown by the Fuller lab at UW to completely protect mice when given as a single intranasal dose 24 h before or after lethal challenge with H3N2 influenza. The Bloom lab at the Fred Hutch has also shown that Flu-Glu has broad specificity to block both H3 and H1 viruses in vitro. Also, Flu-Glue can both capture and detect hemagglutinin in a low cost paper-based diagnostic assay developed in collaboration with the Yager lab at the UW.
How does it work?
As illustrated in Figure 1, researchers designed this potent protein in a two-stage process. They first used Rosetta computational design algorithms to generate a soluble protein that binds with reasonable affinity to the sialic acid binding pocket of the virus’ hemagglutinin protein (HA). This is the site of receptor binding for virus which enables it to grab onto the surface cells and infect them. In a second step, researchers then went on to design a homo-oligomeric trimeric version of the protein that self-assembles to optimally position three the binding proteins to match with near atomic level accuracy the three sialic acid binding pockets in of the self-assembled HA trimer—this is the natural form of HA on the surface of the virus. By perfectly pre-arranging three low affinity HA binders to match three identical pockets on the surface of HA, the team achieved very tight binding to flu HA. The Wilson lab and Ward lab at TSRI confirmed these structures by X-ray crystallography and cryo electron microscopy.
Why is it important?
Many viruses such as Ebola, influenza, respiratory syncytial virus, and others use a trimeric architecture for their cell surface receptor binding proteins. This work proves that protein design can achieve very tight binding to such viral proteins with prophylactic, therapeutic, and diagnostic application. While it is known that antibodies can bind and neutralize viral receptor proteins, their dimeric architectures are not suited to achieve the exquisite affinity and virus blocking ability of of the computationally designed trimeric Flu-Glue.
Researchers in the Baker lab at the Institute for Protein Design, working in collaboration with the Joint Genome Institute, published inScience the solved folds and structures for hundreds of protein families. This “big data” approach to large scale protein structure determination was made possible by a team effort that analyzed billions of gene sequences read out from soil, ocean, and air samples collected around the globe.
Figure 1. Protein Structure Determination from Metagenomic Sequence.
The research has been recognized by numerous opinion leaders and media outlets as an unprecedented breakthrough for protein structure prediction. See articles in The Atlantic, The Economist , Science,GeekWire, and GEN.
How does it work?
As illustrated in Figure 1, the sequencing of DNA from environmental samples produces billions of new protein amino acid sequences. Computer algorithms are used to align the sequences according to their evolutionary history. This allows the discovery of pairs of amino acids that co-evolve. If a change occurs in one amino acid, then a compensatory change is typically observed in another amino acid in the sequence. Co-evolving pairs of amino acids are almost always in close proximity to each other (green and yellow lines) within in the final 3D structure of the protein structure (white backbone).
Why is it important?
With this approach, the team produced reliable models for 622 protein families, and discovered more than 100 new protein folds. In addition to resolving the folding structure of a protein, as shown in Figure 2 co-evolution data can also provide data on the dynamic nature of protein structure including transient contacts, protein-protein contacts, and contacts with ligands. Over time, as more environmental DNA sequence data becomes available, we expect to greatly increase our understanding of protein structure, assembly, and function. In turn, we expect this information to enable the design of new proteins with functions.
Figure 2. Important Protein Contacts Inferred from Co-evoling Amino Acid Pairs.
Sharing data.
The Institute for Protein Design believes in sharing its insights with the rest of the world and we have made publicly available the database of protein structures resolved by these methods.
A few months ago, it was announced that the Institute for Protein Design is one of UW Medicine’s Priorities in their ACCELERATE campaign. We are grateful to have this support not only from UW Medicine, but also from donors who are contributing funds so that we may continue our work. One such gift came from Bruce and Jeannie Nordstrom, whom we’d like to thank for supporting the IPD’s goal to address challenges in medicine, energy, and technology. Click here to read more about the Nordstrom’s support.
An example of computationally designed proteins made of curved beta-sheets and helices forming cavities with different sizes and shapes. (Benjamin Basanta)
The latest paper coming out from the IPD was published today on the Science website. It’s titled “Principles for designing proteins with cavities formed by curved β sheets” with first co-authors Enrique Marcos and Benjamin Basanta, a former and current IPD member, respectively. Other IPD members on the paper include Tamuka Chidyausiku, Gustav Oberdorfer, Daniel-Adriano Silva, Jiayi Dou, and David Baker. Dr. Baker wrote a summary about the publication:
Some of the key functions of the proteins in our bodies and in all living things are to catalyze chemical reactions—speed up the rates by many orders of magnitude-and to sense and respond to small molecules in the body and in the environment. New proteins that catalyze chemical reactions and/or sense and respond to compounds not found in nature would have wide use in medicine and industry.
Another example of computationally designed proteins made of curved beta-sheets and helices forming cavities with different sizes and shapes (Tamuka Chidyausiku)
Computational protein design can in principle be used to generate such new catalysts and receptors, but a major challenge to accomplishing this has been the inability to design proteins with cavities within which the catalysis or small molecule binding can take place. This paper describes a general approach for designing proteins with cavities with tunable size and shape. The method opens the door to design of new catalysts and binding proteins [by generating proteins with appropriately sized and shaped cavities to hold the small molecule and lining the cavity with amino acid functional groups to carry out catalysis and/or binding].
On January 5th, recent IPD spin-out PvP Biologics announced their agreement with Takeda Pharmaceutical Company Limited. The $35 million deal includes an option to acquire PvP at a later point. PvP has released a statement on their website, and the agreement was highly covered by other news sources, which can be found at the following:
We are happy to congratulate Ingrid Swanson Pultz, an IPD Translational Investigator, and Clancey Wolf, a Research Scientist, on PvP Biologics‘ spinout! The news was announced this morning and has been circulated through various outlets. The company, created in 2015, is focusing on advancing KumaMax, a gluten-fighting enzyme that could potentially be taken orally to help those with Celiac disease. To learn more about the announcement, read the full article here.
Seattle-based Arzeda, a computational and synthetic biology company that was spun out from the University of Washington labs of Prof. David Baker, recently announced that its high-throughput, automated pipeline for protein engineering and pathway discovery had been validated by the production of two keystone molecules.
The announcement is a major technical milestone for Arzeda’s approach and their partnership with Amyris, Inc, which is part of a Defense Advanced Research Projects Agency (DARPA) Technology Investment Agreement (TIA).
The two molecules developed with the Arzeda technology are industrially important as dyes, food ingredients and pharmaceutical intermediates, but their current manufacture involves the use of highly toxic and carcinogenic substances such as cyanide and benzene.
“Arzeda’s computational design and synthetic biology technologies for protein design and pathway prediction can now leverage natural fermentation to produce molecules previously only produced through organic chemistry,” said Alexandre Zanghellini, co-founder and CEO of Arzeda. “We can also optimize them in ways not accessible to the synthetic chemist, creating the next generation of products with improved performance. Arzeda’s proprietary technology ushers in a new era of “combinatorial biochemistry” where entirely novel molecules can be made to order. We look forward to working with Amyris to further develop this important technology.”
Prof. Baker, currently the director of the Institute for Protein Design, helped co-found Arzeda, along with Drs. Alexandre Zanghellini, Eric Althoff and Daniela Grabs-Röthlisberger in 2008 to commercialize an innovative computational enzyme design technology based on the Rosetta software, that is used to rapidly make proteins and enzymes with new function.
These proteins and enzymes, and the more efficient and sustainable production pathways developed by Arzeda, have the potential to revolutionize industries ranging from food and pharmaceuticals, to advanced materials and chemicals.
Since its founding, Arzeda has been harnessing the technology to create new enzymes and chemical products that can compete on cost, performance and sustainability. The company has also established partnerships with several leading companies that will help bring these new products to the market.
Dr. Baker and the IPD continue to support Arzeda’s efforts to commercialize these important technologies, and look forward to more great news from Dr. Zanghellini and his team.
More information about Arzeda’s announcement is available at their website.
Published today in Science Philanthropy Alliance, David Baker, Director of the Institute for Protein Design describes how the opportunities for computational protein design are endless — with new research frontiers and a huge variety of practical applications to be explored, from medicine to energy to technology.
This is an exciting time as we are undergoing a technological revolution in protein design—rather than simply tweaking proteins that have come through the evolutionary process, we are becoming able to design new proteins from scratch to solve current challenges.
Computationally Designed Barrel Protein, Image by Possu Huang
Today is Limb Girdle Muscular Dystrophy Day, and the Institute for Protein Design is collaborating with the Jain Foundation and Foldit community to to model the structure of human dysferlin protein (DYSF), an important protein for normal muscle function. Numerous mutations in the gene that encodes DYSF protein are known to be the cause of Limb Girdle Muscular Dystrophy. Our goal is first to first gain a better understanding of the structure of DYSF and then to use the structural information to investigate the effects of LGMD disease mutations on DYSF structure and function.
Read more on DYSF and LGMD at UW’s NewsBeat’s release here and Foldit’s release here.
Small constrained peptides combine the stability of small molecule drugs with the selectivity and potency of antibody-based therapeutics. However, peptide-based therapeutics have largely remained underexplored due to the limited diversity of naturally occurring peptide scaffolds, and a lack of methods to design them rationally. New computational design and wet lab methods developed at the Institute for Protein Design have now opened the door to rational design of awhole new world of hyper-stable drug-like peptide structures.
In an article published in Nature this week, Baker lab / IPD scientists and their collaborators describe the development of computational methods for de novo design of constrained peptides with exceptional stabilities. They used these computational methods to design 18-47 residue constrained peptides with diverse shapes and sizes. The designed peptides presented in the paper cover three broad categories:
2) synthetic disulfide cross-linked peptides with non-canonical sequences, and
3) cyclic peptides with non-canonical backbones and sequences.
Experimentally determined structures for these peptides are nearly identical to their design models.
EHEE Designed Peptide, Visual Illustration by Vikram Mulligan. The molecular surface is shown as a transparent blue shell, and the peptide’s backbone structure is pink. The amino acid’s side chains are white (carbon atoms), blue (nitrogen atoms) and red (oxygen atoms). The crisscrossing bonds that give the peptide its constrained, stable shape are in bright white.
By including D-amino acids (mirror images of the L-amino acids), and thus expanding the palette of building blocks, Baker lab scientists designed peptides in a sequence and structure space sampled rarely by Nature. Indeed, the article describes successful design of a cyclic 2-helix peptide of mix chirality that represents a shape beyond natural secondary- and tertiary structure.
These designed peptides also exhibit exceptional stability to thermal and chemical denaturation, and thus could serve as attractive scaffolds for design of novel peptide-based therapeutics. More broadly, development of this new computational toolkit to precisely design constrained peptides opens the door for “on-demand” development of a new generation of peptide-based therapeutics. See In the Pipeline.
These and other breakthroughs in computational protein design are also covered in a Nature review article by David Baker, Po-Ssu Huang, and Scott E. Boyken entitled “The coming of age of de novo protein design”, part of special supplement on The Protein World.
Illustrations of designed peptides with different configurations of two structures: tightly wound ribbons and flat, arrow-shaped ribbons.
The National Institutes of Health provided partial support for this work through grants P50 AG005136, T32-H600035., GM094597, GM090205, and HHSN272201200025C. Additional funding was provided by The Three Dreamers.
It was a great year for the Institute for Protein Design and we couldn’t have done all of our amazing work without the support from our donors and contributors! Thank you to everyone who helped us, whether through a donation, collaboration, playing Foldit, or otherwise. We’ve filled the IPD Newsletter with all of the progress we’ve made in 2016, so take a look! In the PDF there are links to articles and publications, but many of them can also be found on either this website, or at www.bakerlab.org. Please continue to watch our growth as we head into 2017!
Earlier this month, Baker lab researchers reported the computational design of a hyperstable 60-subunit protein icosahedron in Nature (Hsia et al); icosahedral protein structures are commonly observed in natural biological systems for packaging and transport (e.g. viral capsids). The described design was composed of 60 trimeric protein building blocks that self-assembled in a nanocage.
In new work published today, Baker lab scientists and collaborators have taken this work to an exciting new level by engineering 120-subunit icosahedral nanocages that self-assemble from not one, but two distinct protein components. The new designed proteins are described in the latest issue of Science in a paper entitled “Accurate design of megadalton-scale multi-component icosahedral protein complexes”.
In this paper, former Baker lab graduate student Jacob Bale, Ph.D. and collaborators describe the computational design and experimental characterization of ten two-component protein complexes that self-assemble into nanocages with atomic-level accuracy. These nanocages are the largest designed proteins to date with molecular weights of 1.8-2.8 megadaltons and diameters comparable to small viral capsids. The structures have been confirmed by X-ray crystallography (see figure). The advantage of a multi-component protein complex is the ability to control assembly by mixing individually prepared subunits. The authors show that in vitro mixing of the designed subunits occurs rapidly and enables controlled packaging of negatively charged GFP by introducing positive charges on the interior surfaces of the two copmonents.
The ability to design, with atomic-level precision, these large protein nanostructures that can encapsulate biologically relevant cargo and that can be genetically modified with various functionalities opens up exciting new opportunities for targeted drug delivery and vaccine design. A link to the paper and additional information is below:
Abstract
Nature provides many examples of self- and co-assembling protein-based molecular machines, including icosahedral protein cages that serve as scaffolds, enzymes, and compartments for essential biochemical reactions and icosahedral virus capsids, which encapsidate and protect viral genomes and mediate entry into host cells. Inspired by these natural materials, we report the computational design and experimental characterization of co-assembling two-component 120-subunit icosahedral protein nanostructures with molecular weights (1.8-2.8 MDa) and dimensions (24-40 nm diameter) comparable to small viral capsids. Electron microscopy, SAXS, and X-ray crystallography show that ten designs spanning three distinct icosahedral architectures form materials closely matching the design models. In vitro assembly of independently purified components reveals rapid assembly rates comparable to viral capsids and enables controlled packaging of molecular cargo via charge complementarity. The ability to design megadalton-scale materials with atomic-level accuracy and controllable assembly opens the door to a new generation of genetically programmable protein-based molecular machines.
The Baker lab, in collaboration with Neil King, Trisha Davis and Tamir Gonen’s labs, recently had a paper published in Nature about a stable icosahedral nanocage whose applications could span anywhere from drug delivery to vaccine design! The title is “Design of a hyperstable 60-subunit protein icosahedron” and it was published online June 15, 2016. Yang Hsia, a graduate student in David’s lab, gave an interview to Nature that was put in to a podcast that you can listen to here (called “Protein Football”).
See below for the abstract, or read the whole paper at Nature‘s website here or the Baker lab website here. There is also a great write up of the paper done by Chemical & Engineering News, which you can find here.
The icosahedron is the largest of the Platonic solids, and icosahedral protein structures are widely used in biological systems for packaging and transport1, 2. There has been considerable interest in repurposing such structures3, 4, 5 for applications ranging from targeted delivery to multivalent immunogen presentation. The ability to design proteins that self-assemble into precisely specified, highly ordered icosahedral structures would open the door to a new generation of protein containers with properties custom-tailored to specific applications. Here we describe the computational design of a 25-nanometre icosahedral nanocage that self-assembles from trimeric protein building blocks. The designed protein was produced in Escherichia coli, and found by electron microscopy to assemble into a homogenous population of icosahedral particles nearly identical to the design model. The particles are stable in 6.7 molar guanidine hydrochloride at up to 80 degrees Celsius, and undergo extremely abrupt, but reversible, disassembly between 2 molar and 2.25 molar guanidinium thiocyanate. The icosahedron is robust to genetic fusions: one or two copies of green fluorescent protein (GFP) can be fused to each of the 60 subunits to create highly fluorescent ‘standard candles’ for use in light microscopy, and a designed protein pentamer can be placed in the centre of each of the 20 pentameric faces to modulate the size of the entrance/exit channels of the cage. Such robust and customizable nanocages should have considerable utility in targeted drug delivery6, vaccine design7 and synthetic biology8.
A paper recently published in Science by several members of the IPD, in collaboration with others, entitled “De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity,” discusses designing proteins in a similar way to DNA so that they may be used to engineer structures. Geekwire has written up a great article about the paper; read it here. The abstract is:
General design principles for protein interaction specificity are challenging to extract. In DNA, specificity arises from a limited set of hydrogen-bonding interactions in the core of the double helix to design and build a wide range of shapes. In proteins, specificity arises largely from buried hydrophobic packing complemented by irregular peripheral polar interactions. Protein-based materials have the potential for even greater geometric and chemical diversity, including additional functionality. Here we describe a general approach for designing a wide range of protein oligomers that have interaction specificity determined by modular arrays of extensive hydrogen bond networks. We use the approach to design dimers, trimers, and tetramers consisting of two concentric rings of helices, including previously not seen triangular, square, and supercoiled topologies. X-ray crystallography confirms that the structures overall, and the hydrogen-bond networks in particular, are nearly identical to the design models, and the networks confer interaction specificity in vivo. The ability to design extensive hydrogen-bond networks with atomic accuracy enables the programming of protein interaction specificity for a broad range of synthetic biology applications; more generally, our results demonstrate that, even with the tremendous diversity observed in nature, there are fundamentally new modes of interaction to be discovered in proteins.
Over the weekend, Foldit had its 8th birthday! In celebration, they will be tweeting (@Foldit) fun facts and infographics on their feed. Haven’t heard of the game or tried playing it yet? What better time than now! Click here to learn more and to join an ever-growing community that spans the world. Who knows, maybe you’ll be the person who folds the protein that will help create a way to fight disease!
On Monday, representatives from the IPD spoke on a panel at Xconomy’s EXOME’s Seattle’s Life Science Disruptors 2016 event. It was titled “Proteins Like You’ve Never Seen” and included Lucas Nivon (Cyrus), Ingrid Swanson Putlz (PvP Biologics), Aaron Chevalier (Virvio), and David Baker. The event’s description was as follows:
Seattle is one of the few cities in the world with a dense confluence of biotechnology, medicine, information technology, and public health expertise, and a footprint small enough to encourage intense collaboration between the sectors. From that mix come new ideas, products, and organizations that aim to change the way new therapies are created are created and how people in the U.S. and around the world get their healthcare.
On May 2, we’ll gather at the Fred Hutchinson Cancer Research Center to hear from and talk to some of the most forward-thinking scientists, executives, entrepreneurs, and investors from Seattle and beyond whose work is shaking up entrenched healthcare practices. New drugs. Global initiatives. Better prevention and diagnosis. Deeper analysis. Join us for a healthy dose of thought-provoking conversation.
We are proud to announce that last week, David was named winner of Seattle Business‘ 2016 Leaders in Health Care Awards for “Outstanding Achievement in Delivery of Digital Health.” The award was one of several given out, with categories ranging from “Lifetime Achievement” to “Outstanding Achievement: Health Care Delivery.” Although David was ill and unable to attend the dinner event, Trisha Davis accepted the award in his place. Seattle Business says this about the awards:
As technology improves and Americans spend more on treatments to cure or prevent disease and injury, 2016 is likely to be a challenging year in health care. Doctors, nurses and clinicians are learning to work in new and innovative ways as consumers rely on video consults and their smartphones as diagnostic tools.
The 18 honorees in Seattle Business magazine’s 2016 Leaders in Health Care Awards are up to the challenge. All are champions for change, compassionate visionaries who believe in better patient care. They are harnessing technology, tackling the unknown and solving scientific puzzles — all in the name of promoting health and lowering costs.
They are forward thinkers who excel in the delicate balancing act involving the health, lives and resources of consumers. Congratulations to this year’s honorees for their commitment to ensuring Washington’s health care industry remains at the forefront of worldwide achievement.
There is also a separate article about David here.
Additionally, back in January, David was listed amongst Thomas Reuters’ list of “World’s Most Influential Scientific Minds” for 2015. He was one among 27 UW faculty members that made the list, which is determined by most citations by peers for various fields. You can read the UWToday article here. David was selected for biology and biochemistry.
On February 18th, WRF FellowFranziska Seeger gave a talk about proteins at Ignite Seattle. Presenters have 5 minutes to speak to the audience and deliver whatever point they are trying to get across. They also cannot control their slides, so they have to have their timing perfect. Talk about pressure! However, we believe that Franziska did an excellent job explaining “Molecular Machines and Designer Drugs.” Please see below to watch her presentation.
On February 9th, Arzeda, and Mitsubishi Rayon Ltd announced that they would “collaborate on development of new processes for industrial chemicals production.” Read more about it on Mitsubishi Rayon’s website, here, or on Arzeda’s site, here.
Arzeda is an IPD spin-out, of which David Baker is a co-founder and Scientific Advisory Board Member.
Today at 11am, the paper titled “A Computationally Designed Hemagglutinin Stem-Binding Protein Provides In Vivo Protection from Influenza Independent of a Host Immune Response” was published to the PLOS Pathogen website. This paper was contributed to by several IPD members, including Aaron Chevalier, Jorgen Nelson, Lance Stewart, Lauren Carter, and David Baker. The research was performed in collaboration with colleagues in Deborah Fuller’s lab at UW’s Department of Microbiology. You can find the paper here. Scroll down for the official press release.
FluBinder Rendered by Vikram Mulligan, PhD
Fighting Flu with Designer Drugs: A New Compound Given Before or After Exposure Fends Off Different Influenza Strains
A study published on February 4th in PLOS Pathogens reports that a new antiviral drug protects mice against a range of influenza virus strains. The compound seems to act superior to Oseltamivir (Tamiflu) and independent of the host immune response.
Influenza viruses under the microscope look a bit like balls covered with spikes. The spikes are actually two different proteins, hemagglutinin (HA) and neuraminidase (NA). Both proteins consist of an inner stem region (which doesn’t differ much between flu strains), and a highly variable outer blob. The individual variants fall into designated groups, and this is how flu strains are categorized (for example as H1N1, or H3N5).
Ongoing mutations that change the HA and NA blobs are the reason why flu vaccines differ from season to season; they are based on researchers’ best guesses of what next year’s prominent strains will look like. And dangerous pandemic strains often have radically new blobs against which existing immunity is limited.
In the search for drugs that act broadly against different influenza strains, researchers had previously shown that antibodies against the HA stem region can prevent influenza infection. Such antibodies are protective, at least in part, because they activate the host immune response which then destroys the whole HA/antibody complex. The approach, then, depends on a fully functional immune system—which is not present in infants, the elderly, or immune-compromised individuals.
Inspired by the earlier work, Deborah Fuller from the University of Washington in Seattle, USA, who is interested in developing influenza drugs and vaccines, teamed up with David Baker, also at the University of Washington, who is an expert in computational protein design. Together with colleagues, they set out to design small molecules that—like the protective antibodies—bind to the HA stem, and to test whether these small molecules can protect against influenza infection. Designed to mimic antibodies, the small molecules bind the virus in a similar manner. However, because they don’t engage the immune system the way antibodies do, and because of questions of stability and potency, it was not clear whether they would be able to prevent infection in animals, or eventually, in humans.
Before testing their molecules in animals, the researchers optimized their favorite small molecule candidate by systematically generating thousands of versions and testing how tightly they bound HA stems from seven different influenza strains. As they predicted, the resulting molecule, called HB36.6, protected cells against influenza virus infection in vitro (i.e., in test tubes).
The researchers next tested HB36.6 in “challenge experiments” in mice. They gave mice a single intranasal dose of the drug and 2 hours, 24 hours, or 48 hours later injected them with a normally lethal dose of influenza virus. This one-time HB36.6 treatment, when given up to 48 hours before the challenge, conveyed complete protection: All of the treated mice survived and had little weight loss, whereas all untreated control mice died after losing a third of their body weight or more. Intranasal HB36.6 was also able to protect mice after they had been exposed to flu virus, when administered either as a single dose within a day after exposure, or when it was given daily for four days starting 24 hours after exposure.
This protection does not depend on an intact host immune response. When the researchers repeated the challenge experiments in two different immune-deficient mouse strains, they found that HB36.6 can protect these mice as well.
Comparing HB36.6 with Oseltamivir, the researchers found that a single dose of HB36.6 provided better protection than 10 doses (twice daily for 5 days) of Oseltamivir. Furthermore, when they gave a low dose of HB36.6 post-infection (which by itself was not able to afford full protection) together with twice-daily doses of Oseltamivir, all the mice survived, indicating a synergistic effect when the two antiviral drugs are combined.
Their results, the researchers conclude, “show that computationally designed proteins have potent anti-viral efficacy in vivo and suggests promise for development of a new class of HA stem-targeted antivirals for both therapeutic and prophylactic protection against seasonal and emerging strains of influenza”.
Recently, UW Medicine Pulse released a podcast featuring none other than our very own Ingrid Swanson Pultz! They talked to her about KumaMax and how it would help those with Celiac’s disease. Go here to see their post on it and listen to the podcast!
Would you like to contribute to her research? Please go here to give a gift that will help further her work.
Custom design with atomic level accuracy enables researchers to craft a whole new world of proteins
Naturally occurring proteins are the nanoscale machines that carry out essentially all of the critical functions in living things.
While it has been known for over 40 years that the sequence of amino acids completely determines the shape of the protein, it has been very challenging to predict from the amino acid sequence of the protein its three-dimensional structure, and conversely, to come up with brand new amino acid sequences which fold up into hitherto unseen structures.
Over the past months, scientists at the Institute for Protein Design at the University of Washington and the Fred Hutch, along with colleagues at other institutions, have reported advances in two long-standing problem areas related to the construction of new proteins from scratch.
“It has been a watershed year for protein structure prediction and design,” said UW Medicine researcher David Baker, a University of Washington professor of biochemistry, Howard Hughes Medical Institute investigator and head of the Institute for Protein Design.
The protein structure problem is about figuring out how a protein’s chemical makeup predetermines its molecular structure, and in turn, its biological role. UW researchers have developed powerful new algorithms using co-evolution data from DNA sequences to make unprecedented highly accurate blind ‘ab initio’ structure predictions of large proteins (>200 amino acids in length). This has opened the door to accurate prediction of the structures for hundreds of thousands of newly discovered proteins in the ocean, soil, and gut microbiome.
Equally difficult is the second problem, which is designing amino acid sequences that will fold into brand new protein structures. Breakthroughs demonstrate that it is now possible to make brand new amino acid sequences with exacting precision for folds inspired by the natural world; and more importantly to make amino acid sequences from scratch for totally novel unknown folds, far surpassing what is predicted to occur in natural proteins.
The new proteins are designed with the help of volunteers around the world participating in the Rosetta@Home distributed computing project. The designed amino acid sequences are encoded in synthetic genes, the proteins are produced in the laboratory, and their structures determined with X-ray crystallography. The computer models in almost all cases match the experimentally determined crystal structures with near atomic level accuracy.
Researchers report new protein designs for barrels, sheets, rings, and screws –all with near atomic level accuracy. This builds on previous reports of designed protein cubes and spheres; providing proof that it is possible to make a totally new class of protein materials.
With these advances in both protein structure prediction and molecular design, Institute for Protein Design researchers hope to build a new world of proteins with exact specifications for performing critically needed tasks in medical, environmental and industrial arenas.
Examples of their goals are nanoscale tools that:
boost the immune response against HIV and other recalcitrant viruses
block the flu virus so that it can’t infect cells
deliver drugs to cancer cells with precision and reduced side effects
stop allergens from causing symptoms
neutralize deposits, called amyloids, thought to damage vital tissues in Alzheimer’s disease
mop up medications in the body as an antidote
fulfill other diagnostic, therapeutic, and clean energy needs
Just as the manufacturing industry was revolutionized by creating interchangeable parts designed to precise specifications, custom designed protein modules with the right twist, turns, and connections for their modular assembly is a bold new direction for biotechnology.
Results providing proof of this possible future have been reported in recent months by researchers the UW Institute for Protein Design in collaboration with researchers at the Fred Hutch, Max Planck Institute for Developmental Biology, Janelia Research Campus, and the Institute for Molecular Science in Japan.
Evolution offers clues to shaping proteins: The function of many proteins tends to stay the same across species, even after their amino acid sequences have changed over billions of years of evolution. Locating co-evolved pairs of amino acids helps calculate their proximity when the molecule folds. UW graduate student Sergey Ovchinnikov applied this co-evolution DNA sequence analysis in an E-Life paper published on September 3, 2015 entitled “Large-scale determination of previously unsolved protein structures using evolutionary information” that illuminated for the first time the structures of 58 families of proteins containing hundreds of thousands of additional structurally related family members.
“This achievement was a grand slam home run in the history of protein structure prediction,” said Baker.
Modular construction of proteins with repeating motifs: Proteins composed of repeated modules, similar to interlocking Lego® blocks, are common in the natural world. Two papers published in the December 16 issue of Nature entitled, “Exploring the repeat protein universe through computational protein design,” and “Rational design of alpha-helical tandem repeat proteins with closed architectures,” shows that existing repeat proteins occupy only a small fraction of the available space, and that it is possible to design totally new proteins with precisely specified geometries that go far beyond what nature has achieved. The work was led by postdoctoral fellows TJ Brunette, Fabio Parmeggiani and Po-Ssu Huang in the lab of David Baker at the University of Washington Institute for Protein Design and Lindsey Doyle and Phil Bradley at the Fred Hutchinson Cancer Research Institute in Seattle.
Barrel-fold design: , Baker lab postdoctoral fellow Po-Ssu Huang, together with Birte Höcker at the Max Planck Institute for Developmental Biology (Tübingen, Germany) discovered the critical but elusive design principles for a barrel-shaped fold underpinning many natural enzyme molecules. The custom designed barrels folds were built at the Institute for Protein Design and reported on November 23, 2015 in the Nature Chemical Biology paper, “De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy.” This breakthrough has opened the door for bioengineers to generate totally new enzymes that speed up chemical reactions by positioning smaller molecules in custom barrel compartments.
Self-assembling apparatus: Naturally occurring ordered protein arrays along a flat plane are found in bacteria, the heart, and other muscles. Overcoming protein interaction complexities, researchers at UW Institute for Protein Design and the Janelia Research Campus of the Howard Hughes Medical Institute succeeded in programming proteins to self-assemble into novel symmetric, 2-dimensional sheets of protein lattice patterns. UW graduate student Shane Gonen in the Baker lab together with his brother Tamir Gonen at Janelia described their work in the June 19, 2015 issue of Science, “Design of ordered two-dimensional arrays mediated by non-covalent protein-protein interfaces.” This research has application in the design self-assembling protein nanomaterials, especially those that could serve as efficient sensors or light harvesters.
Precision sculpting: Protein designers are continuously refining the principles for fashioning ideal protein structures. The latest paper in the October 6, 2015 Proceedings of the National Academy of Sciences, “Control over overall shape and size in de novo designed proteins” further explains methods for systematically varying protein architecture inspired by nature. Such finesse is needed in optimizing designed proteins to take on exact shapes to perform specified functions. This work has been led by Baker lab graduate student Yu-Ru Lin in collaboration with Nobuyasu Koga at the Institute for Molecular Science in Japan.
Funding Sources:
The Institute of Protein Design has been funded by several federal agencies, including National Institutes of Health, U.S. Department of Energy, National Science Foundation, U.S. Defense Threat Reduction Agency, and U.S. Air Force Office of Scientific Research, the Washington Research Foundation, the Life Sciences Discovery Fund, as well as through private support.
The Institute also depends on a cadre of citizen scientists around the world who volunteer their personal and computer time for protein folding prediction studies through Rosetta@home and the multi-player on-line protein folding game Foldit.
A similar story was also published in UW Health Science’s Newsbeat. Read it here.
In the early 1990s, researchers in the field of protein structure prediction were challenged by the problem of how to impartially judge the accuracy of prediction algorithms. This realization led the protein structure prediction the community to start the Critical Assessment of protein Structure Prediction (CASP), a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. In each CASP, an independent scientific advisory board solicits other researchers to submit experimentally verified, but unpublished, 3D protein structures to CASP. The linear amino acid sequences of these proteins are then provided to structure prediction researchers, who each have an equal and limited amount of time to submit final structure predictions to the CASP advisory board. The submitted structure predictions are then compared to the experimentally verified structures using the same metrics for all CASP contributors. Even though the primary goal of CASP is to help advance methods for identifying protein 3D structure given only its linear amino acid sequence, many view the experiment more as a “world championship” in protein structure prediction.
Over a 16 year period (CASP3-11), the Baker lab has consistently achieved top performance in the hardest category of structure prediction; the “Twilight Zone” where the linear amino acid sequence of the protein shares no discernable relation to any publicly available 3D structure. In 2014 this culminated in our highly accurate blind structure predictions of two large proteins each >200 amino acids in length. Our methods involve using DNA sequence information to help us predict the 3D structures of proteins.
We recently published these results in E-life, and the results are getting significant attention.
IPD Translational Investigator, Dr. Ingrid Pultz, published a paper in JACS this month titled ‘Engineering of Kuma030: A Gliadin Peptidase That Rapidly Degrades Immunogenic Gliadin Peptides in Gastric Conditions‘. Using Rosetta to redesign the active site of the gliadin protease KumaMax – an enzyme computationally designed to break down gluten in the stomach – Dr. Pultz and collaborators show that the new variant Kuma030 degrades >99% of the gluten peptide that triggers inflammation in celiac disease patients. This work brings us even closer to arriving at an oral therapeutic for celiac disease.
IN THE NEWS
Dr. Pultz was interviewed by MyNorthwest.com on her work developing a pill that celiac patients can take before consuming gluten. Read and hear more at the link:
The IPD hosted its second Scientific Council meeting this month, chaired by David Urdal, PhD, MS. The council is made up of UW and Fred Hutch faculty from a variety of departments (Oncology, Genome Sciences, Immunology, Allergy & Infectious Diseases, Biochemistry, and Pharmacology). The goal of the IPD Scientific Council is threefold:
1. Identify new opportunities, targets, and applications to which protein design can be applied
2. To strategize on how best to balance core technology development with translational projects of value today and translational projects with important impacts 5 to 10 years down the road
3. Provide feedback on current projects
IPD Director Dr. David Baker was the Keynote speaker at the 13th Annual NanoDDS (International Nanomedicine and Drug Delivery) symposium, held at the UW this year. Dr. Baker gave a talk entitled ‘Engineering Protein Nanocarriers: Deisgn of protein interaction inhibitors and self-assembling nanocages’.
Dr. Baker also spoke on a panel at the Washington State Academy of Sciences 8th Annual Symposium on “Accelerating Science’s Impact: Translating Discoveries Into Solutions”. Held at the Museum of Flight, the panel was moderated by UW CoMotion Executive Director Vikram Jandhlaya and panelists discussed various topics under the theme of “Translational Science for Health and Disease Barriers and Solutions”.
Dinner with brilliant David Baker after wonderful WSAS panel:managed to get the panelists to argue a bit too-fun! #fbpic.twitter.com/8wrMLrczhh
Following the groundbreaking 2014 Nature paper describing the development of a computational method to design multi-component coassembling protein nanoparticles, comes a publication in Protein Sciencefrom Baker lab graduate student Jacob Bale and collaborators. Titled “Structure of a designed tetrahedral protein assembly variant engineered to have improved soluble expression“, the paper reports a variant of a previously low yielding tetrahedral designed material for which structure determination was difficult. The new variant described in the paper had a much improved yield after redesign and the structure obtained agreed with the computational model with high atomic-level accuracy. The methods used here to improve soluble protein yield will be generally applicable to improving the yield of many designed protein nanomaterials.
INSTITUTE
Congratulations to newly minted PhDs and graduates of the Baker lab Dr. Shawn Yu and Dr. Ray Wang! Both defended their dissertations this month. Dr. Yu gave a talk on “Computational design of interleukin-2 mimetics” and Dr. Wang spoke about “Protein structure determination from cryoEM density maps”. We wish them the best of the luck in their next steps!
The annual RosettaCONmeeting was held July 29-Aug1 at the beautiful Sleeping Lady Mountain Resort in Leavenworth, WA. Many IPD scientists attended the conference, heard talks from researchers in Rosetta labs across the country, presented posters on their own research, and socialized with the larger Rosetta community.
In an article out in Structure, Baker lab postdoc Dr. Hahnbeom Park in collaboration with IPD Assistant Professor Frank DiMaio investigate the origin of protein structure refinement from structural averaging at the residue level. Structure refinement has long been a challenge in the field of protein structure prediction and it aims to improve homology models to the level of experimentally determined structures. Their studies and conclusions can be found in a paper titled “The Origin of Consistent Protein Structure Refinement from Structural Averaging“.
INSTITUTE
A big welcome to new UW Biochemistry and IPD Assistant Professor Dr. Liangcai Gu! Dr. Gu is also an Adjunct Assistant Professor of Genome Sciences. Dr. Gu’s lab is interested in using quantitative protein interaction profiling to understand molecular recognition and guide computation protein design. We are very excited to have Dr. Gu as part of our protein design team!
The IPD also welcomes visiting scholar Wally Novak, VISIT intern Can Li, and visiting research scientist Maziar Ardejani!
David Baker gave a talk at the Institute for Systems Biology in Seattle titled “Post-Evolutionary Biology: Design of novel protein structures, functions, and assemblies”.
Dr. Roman Jerala, from the National Institute of Chemistry in Ljubljana, Slovenia visited the IPD and gave a talk on “Design of modular topological folds”.
June was an exciting month for the IPD Translational Investigator program! The IPD launched its very first spinout – Cyrus Biotechnology. Founded by former IPD postdocs Drs. Lucas Nivon and Yifan Song, and former Baker lab graduate student Dr. Javier Castellanos, Cyrus aims to pursue commercialization of an innovative user friendly software as a service (SaaS) cloud computing solution for distribution of the powerful “Rosetta” protein structure prediction and design algorithms. Read the IPD’s full announcement here and explore the Cyrus website at www.cyrusbio.com for more information.
Congratulations to Baker lab graduate student Austin Day on his successful PhD thesis defense!
Translational Investigators along with Dr. Lance Stewart, IPD Sr. Director of Strategy, shared advancements in IPD research at the widely attended WBBA Life Science Innovation Northwest (LSINW) this month. LSINW “connect world-class industry leaders and showcases the Pacific Northwest as a global center for life science advancement”
Cyrus Bio founder Dr. Lucas Nivon and UW CoMotion’s Josh Pan at LSINW 2015
Translational Investigator Dr. Ingrid Swanson Pultz before her presentation on an oral therapeutic for celiac disease at LSINW 2015
We describe a general approach to designing two-dimensional (2D) protein arrays mediated by noncovalent protein-protein interfaces. Protein homo-oligomers are placed into one of the seventeen 2D layer groups, the degrees of freedom of the lattice are sampled to identify configurations with shape-complementary interacting surfaces, and the interaction energy is minimized using sequence design calculations. We used the method to design proteins that self-assemble into layer groups P 3 2 1, P 4 2(1) 2, and P 6. Projection maps of micrometer-scale arrays, assembled both in vitro and in vivo, are consistent with the design models and display the target layer group symmetry. Such programmable 2D protein lattices should enable new approaches to structure determination, sensing, and nanomaterial engineering.
Shown is a 2D array with P 6 symmetry. (Left) the P 6 lattice has two degrees of freedom available for sampling. Sixfolds are represented by hexagons. (Middle) Computationally designed 2D array. (Right) Electron microscopy of designed P 6 array.
Today, we are announcing that Cyrus Biotechnology, has been successfully launched from the UW Institute for Protein Design (IPD) to pursue commercialization of an innovative user friendly software as a service (SaaS) cloud computing solution for distribution of the powerful “Rosetta” protein structure prediction and design algorithms.
“Cyrus is the first new IPD spin out, graduating from the IPD’s Translational Research program in less than 12 months from inception,” said Dr. David Baker, Director of the IPD. Cyrus is building a custom-designed protein modeling and design GUI, automating complex user procedures and deploying on the cloud to customers in Pharma, Biotech and the Chemical industries.
The company’s name was inspired by Cyrus Levinthal’s famous paradox, that most small proteins fold spontaneously on short time scales of less than a millisecond, despite there being are a very large number of degrees of freedom in an unfolded protein chain of amino acids, leading to an astronomical number of possible conformations that may need to be sampled before folding into a low energy conformation. The Rosetta suite of algorithms that originated over 15 years ago at the UW, now with a team of over 100+ programmers contributing to the Rosetta Commons, in many cases solves Levinthal’s paradox.
The IPD initiated its Translational Investigator program in April 2014 with $1.4 M in Opportunity Grant funding from the Life Science Discovery Fund (LSDF) and matching commitments of $5.2 M from the UW, Washington Research Foundation, and the generosity of local philanthropists. In addition to the Cyrus project, IPD Translational Investigator research programs include an oral therapeutic for celiac disease, flu viral therapeutics and diagnostics, and others in development. The Translational Investigator research programs enable talented postdoctoral fellows and graduate students to apply protein design research towards the challenge of real world problems, transitioning protein design research discoveries into Seattle area startup companies. The combination of LSDF Opportunity Grant funding with philanthropist matching funds is being applied to several ongoing translational projects.
“With our LSDF funding, matching grants from philanthropists, and funding from the National Science Foundation I-Corps program, the IPD was able to support the Cyrus co-founder team from the inception of concept to launch of a new Seattle area startup company, all in collaboration with CoMotion and several stakeholders,” said Dr. Lance Stewart, Senior Director of Strategy for the IPD, who also serves as a mentor and advisor to the Translational Investigators.
The Cyrus translational project was conceived in April 2014 by Dr. Lucas Nivon, then a postdoctoral fellow in the Baker lab, together with two other Baker lab members, Dr. Yifan Song who at the time was a postdoc/acting instructor and Javier Castellanos who at the time was in his final year as a graduate student. Shortly after having conceived of the Cyrus concept, the co-founding team incorporated Cyrus and secured meetings with the W-Fund and the local WINGS angel investor network; together these investors have capitalized the IPD spin out with $850K in seed financing.
Throughout their 12 month incubation period, the Cyrus team completed the NSF I-Corps program, developed a prototype of the Cyrus platform, and worked with the UW CoMotion technology transfer office (Dr. Jennifer McCullar and Dr. Dennis Hanson, Senior Technology Managers) and the Rosetta Commons to secure the required intellectual property licensures needed to pursue the Cyrus business model.
“Dr. Jennifer McCullar was instrumental in working with RosettaCommons and CoMotion to facilitate our license negotiations,” said Dr. Lucas Nivon, CEO and Co-Founder of Cyrus. “Our company intends to change the way drugs are discovered by biotechnology and pharmaceutical companies, from a wet lab intensive effort to a computer-aided-design (CAD) engineering task enabled by cloud based Rosetta drug design and discovery.”
The last day of work at the UW for Lucas, Yifan, and Javier was May 1, 2015, when they transitioned to the Cyrus facilities in WeWork at South Lake Union. We wish them the very best.
The May IPD News Roundup covers a new Science paper from IPD Assistant Prof Frank DiMaio, a KOMO News interview with Translational Investigator Ingrid Swanson Pultz on celiac disease, and much more! At the link.
Extremophiles, microorganisms thriving in extreme environmental conditions, must have proteins and nucleic acids that are stable at extremes of temperature and pH. The nonenveloped, rod-shaped virus SIRV2 (Sulfolobus islandicus rod-shaped virus 2) infects the hyperthermophilic acidophile Sulfolobus islandicus, which lives at 80°C and pH 3. We have used cryo–electron microscopy to generate a three-dimensional reconstruction of the SIRV2 virion at ~4 angstrom resolution, which revealed a previously unknown form of virion organization. Although almost half of the capsid protein is unstructured in solution, this unstructured region folds in the virion into a single extended a helix that wraps around the DNA. The DNA is entirely in the A-form, which suggests a common mechanism with bacterial spores for protecting DNA in the most adverse environments.
The SIRV2 protein dimer helices fully encapsulate the DNA. (A) Three asymmetric units of the virion are shown, illustrating how the N-terminal helices wrap around the DNA, forming antiparallel helix-helix packing. (B) Side view. (C) Surface view of the protein (using a 1.4 Å probe radius). (D) The right-handed solenoidal supercoiling of the DNA, with three turns shown.
The March 2015 IPD news roundup is here! A new publication in PNAS on a completely novel metabolic pathway made via protein design and more. Read about it at the link.
This month’s roundup features an interview and two papers from IPD Assistant Professor Frank DiMaio as well as a new Science paper from the Baker lab on trapping transition states using protein design. Read more at the link.
A little late this month but it’s here!!! The January news roundup talks about a new Nature journal paper from the Baker lab, the second Mini Symposium, and much more. Follow the link and read more!
The Institute for Protein Design is seeking an MBA student to work with Institute leadership to collect market data on small molecule therapeutic targets for protein design. More information about this exciting independent study opportunity can be found on our Employment page here.
The Institute for Protein Design is pleased to announce its new advisory board! Follow the link to our news release for more information on our board members.
Our next Mini Symposium will be hosted on Jan 20 2015 and will feature Drs. Bill DeGrado (UCSF) and Gevorg Grigoryan (Dartmouth) speaking on “New Approaches to Protein Design”. More information at this link.
We wrapped up 2014 with a bang – introducing a new logo, getting a major award for celiac disease research, and successfully hosting our first Mini Symposium! Follow the link to read more.
Please make a tax deductible donation at thisLINK.
IPD Translational Investigator Dr. Ingrid Swanson Pultz was awarded a Matching Grant award of $250K from the Life Sciences Discovery Fund (LSDF) for her project ‘In vivo assessment of an oral therapeutic for celiac disease‘!
We need to raise an additional $74K to make the full match.
Learn more about this project by watching this short video.
The goal of this LSDF funded research is to assess the efficacy, safety, and optimal dosing of KumaMax and its variants as an oral enzyme therapy for celiac disease.
KumaMax is the winner of the 2013 Innovation Award at the UW. KumaMax is a computationally designed enzyme which efficiently breaks down gluten in the stomach before it reaches the small intestine where it can cause inflammation in celiac disease patients.
The LSDF Matching Grant has the requirement that the UW must raise an additional 1:1 match of $250K to support this innovative project.
We need your support ! The Institute for Protein Design has received $176K in matching funds from generous philanthropists to support this work.
Every $ counts. We thank everyone for their generous support.
Staff and scientists of the IPD and Foldit volunteered at Pacific Science Center’s Life Sciences Research Weekend this month – teaching budding scientists about the awesomeness of protein folding! Pics from the event and more Institute news at the link.
The UW Institute of Protein Design (IPD) presents a Mini Symposium on “Sweet Spots for Designed Proteins as Therapeutics” on Weds Dec 10 at 9 AM in HSB D-209. Follow the link to get more details! We hope to see you there!
A new paper is out in this week’s issue of Science entitled High thermodynamic stability of parametrically designed helical bundles. Using novel computational design methods, extremely stable helical bundles can be custom designed with fine-tuned structural geometries for a number of applications. Read more about this exciting work at this link.
We awarded our first round of WRF Innovation Postdoctoral fellowships this month! Follow the link to learn about our new fellows and to catch up on the research ongoing at the IPD.
The Foldit community has been leading the charge in the computational design of proteins to bind Ebola. Learn more about this work and other ongoing Institute news at the link.
Learn how the IPD and Translational Investigator Dr. Ingrid Swanson Pultz are developing Kumamax, an oral therapeutic candidate for celiac disease. A slide presentation and more information at the link.
Read a letter from Institute for Protein Design Director Dr. David Baker summarizing the exciting accomplishments and progress made at the IPD in the past year.
What if scientists could design a completely new protein that is precision-tuned to bind and inhibit cancer-causing proteins in the body? Collaborating scientists at the UW Institute for Protein Design (IPD) and Molecular Engineering and Sciences Institute (MolES) have made this idea a reality with the designed protein BINDI. BINDI (BHRF1-INhibiting Design acting Intracellularly) is a completely novel protein, based on a new protein scaffold not found in nature, and designed to bind BHRF1, a protein encoded by the Epstein-Barr virus (EBV) which is responsible for disregulating cell growth towards a cancerous state. Learn more here.
A new paper is out in the June 5 issue of Nature entitled Accurate design of co-assembling multi-component protein nanomaterials. Scientists at the Institute for Protein Design (IPD), in collaboration with researchers at UCLA and HHMI, have built upon their previous work constructing single-component protein nanocages and can now design and build self-assembling protein nanomaterials made up of multiple components with near atomic-level accuracy. Learn more about this innovative work at this link.
In a recent PNAS paper entitled “Removing T-cell epitopes with computational protein design”, IPD researchers combine machine learning with computational protein design to demonstrate immune silencing of protein targets. This deimmunization has the potential to reduce or eliminate immunogenicity of protein therapeutics. Learn more at this link.
A recent Nature issue exposed the dismaying fact that many women are deterred from pursuing a career in science, especially at the highest levels (postdoctoral positions, faculty position, scientific advisory boards to start up companies, etc). To talk about this significant gender gap in science and the issues female scientists face, Baker lab members participated in an informal lunch discussion to determine what specific steps could be taken as a group to encourage and promote women within our own scientific community. Learn more at this link.
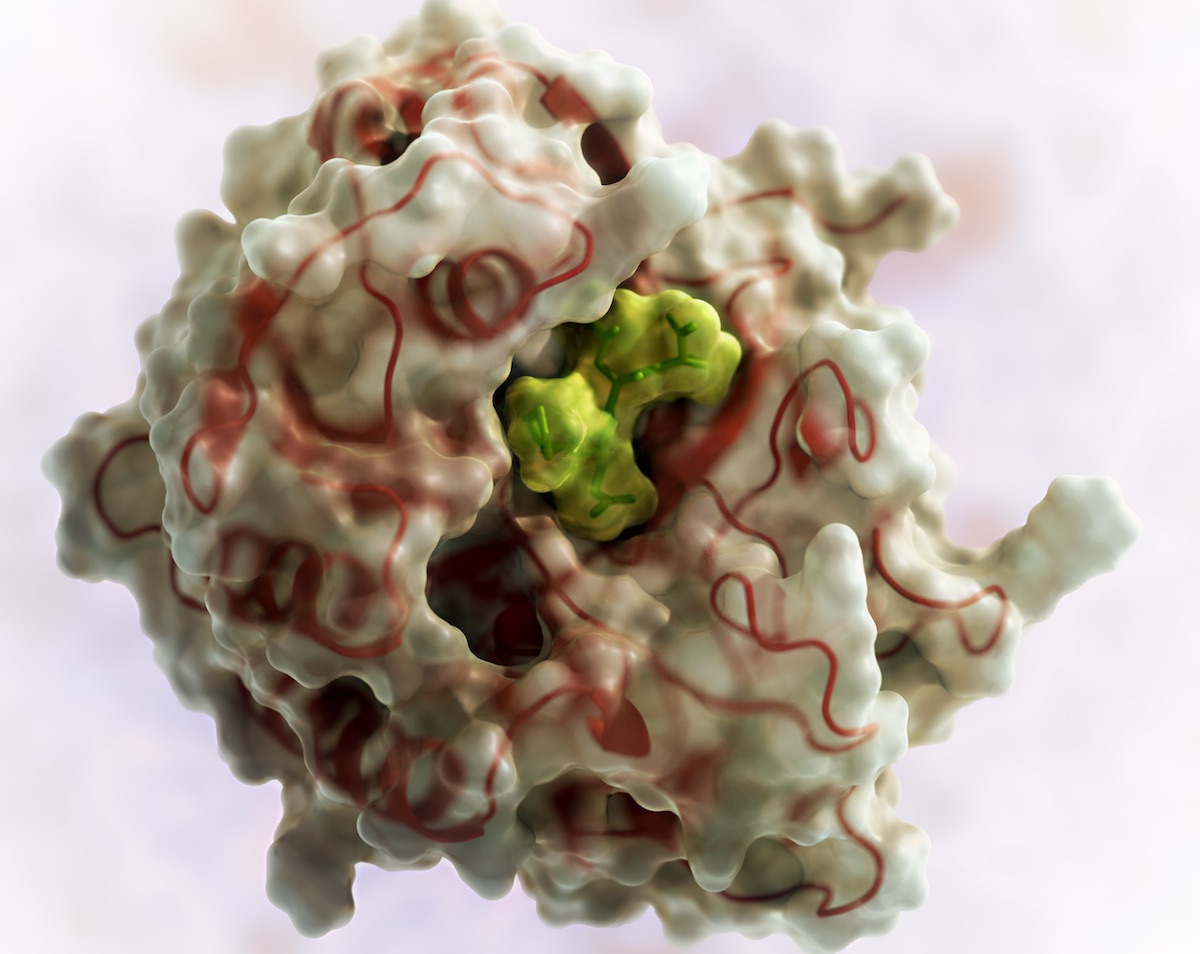
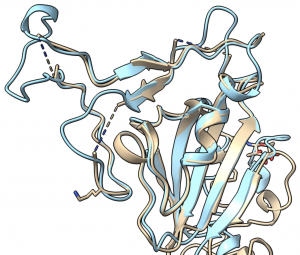
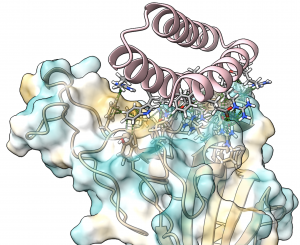
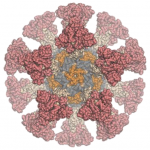

 “Spinning life science innovations out of research institutions requires expertise and funding that is hard to source in the academic environment,” adds
“Spinning life science innovations out of research institutions requires expertise and funding that is hard to source in the academic environment,” adds 

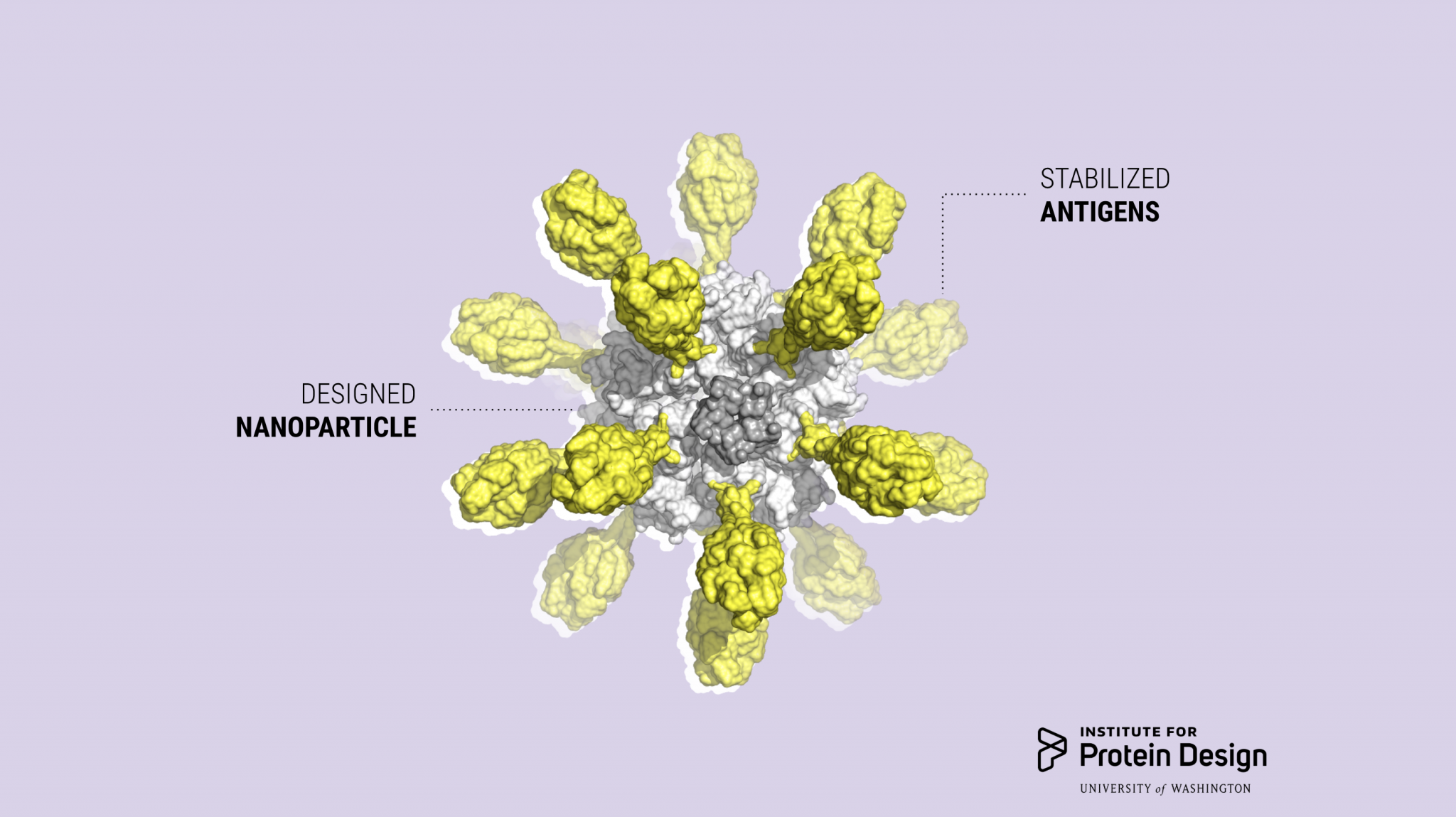
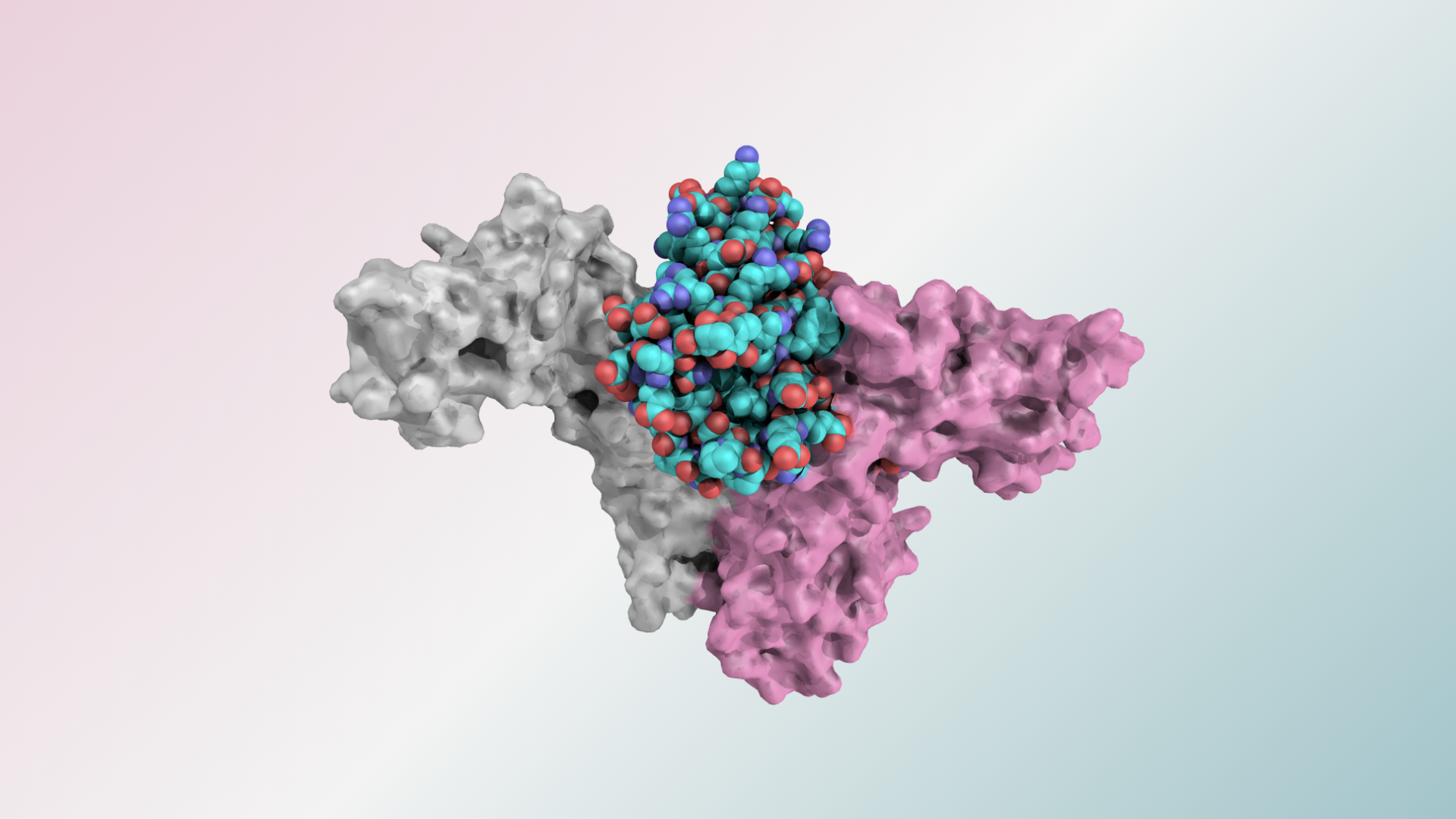


 The idea for LOCKR grew out of a
The idea for LOCKR grew out of a 
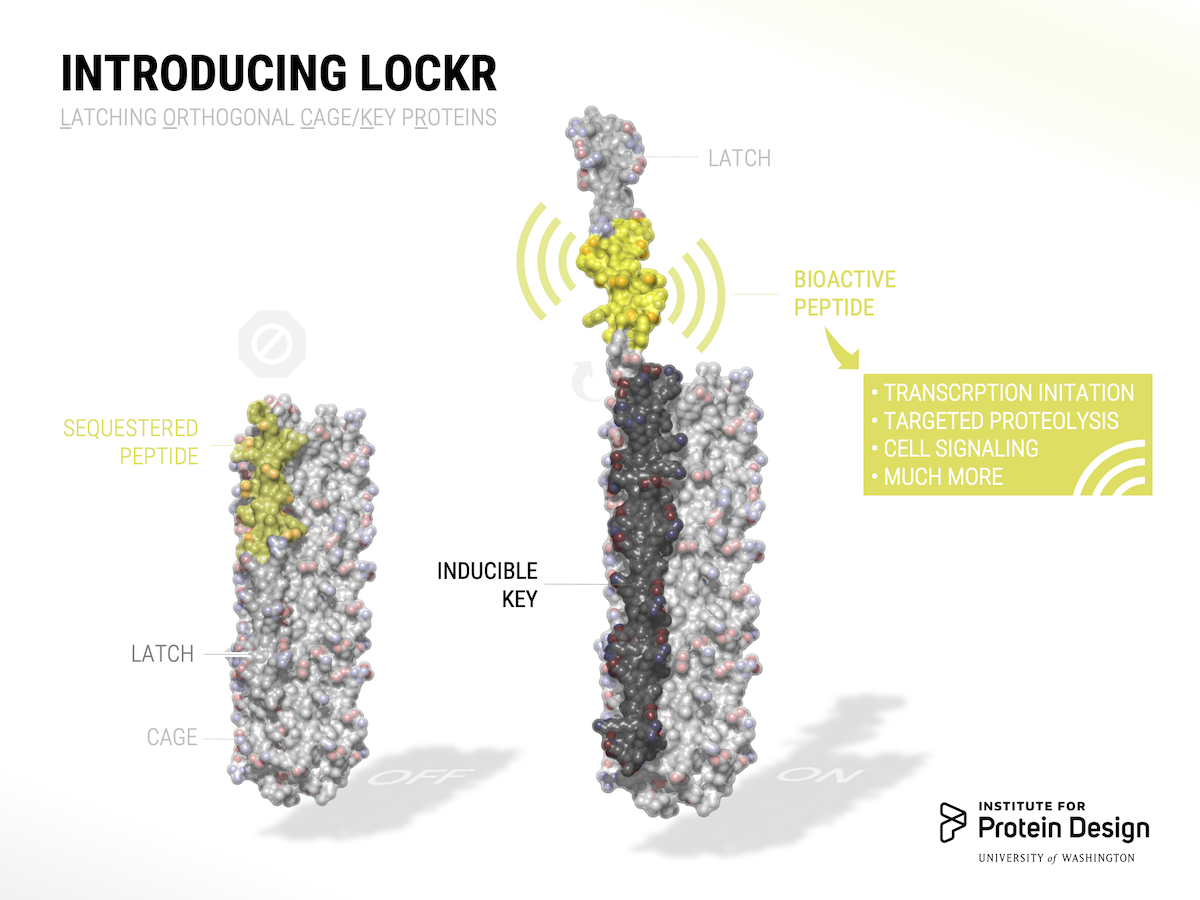


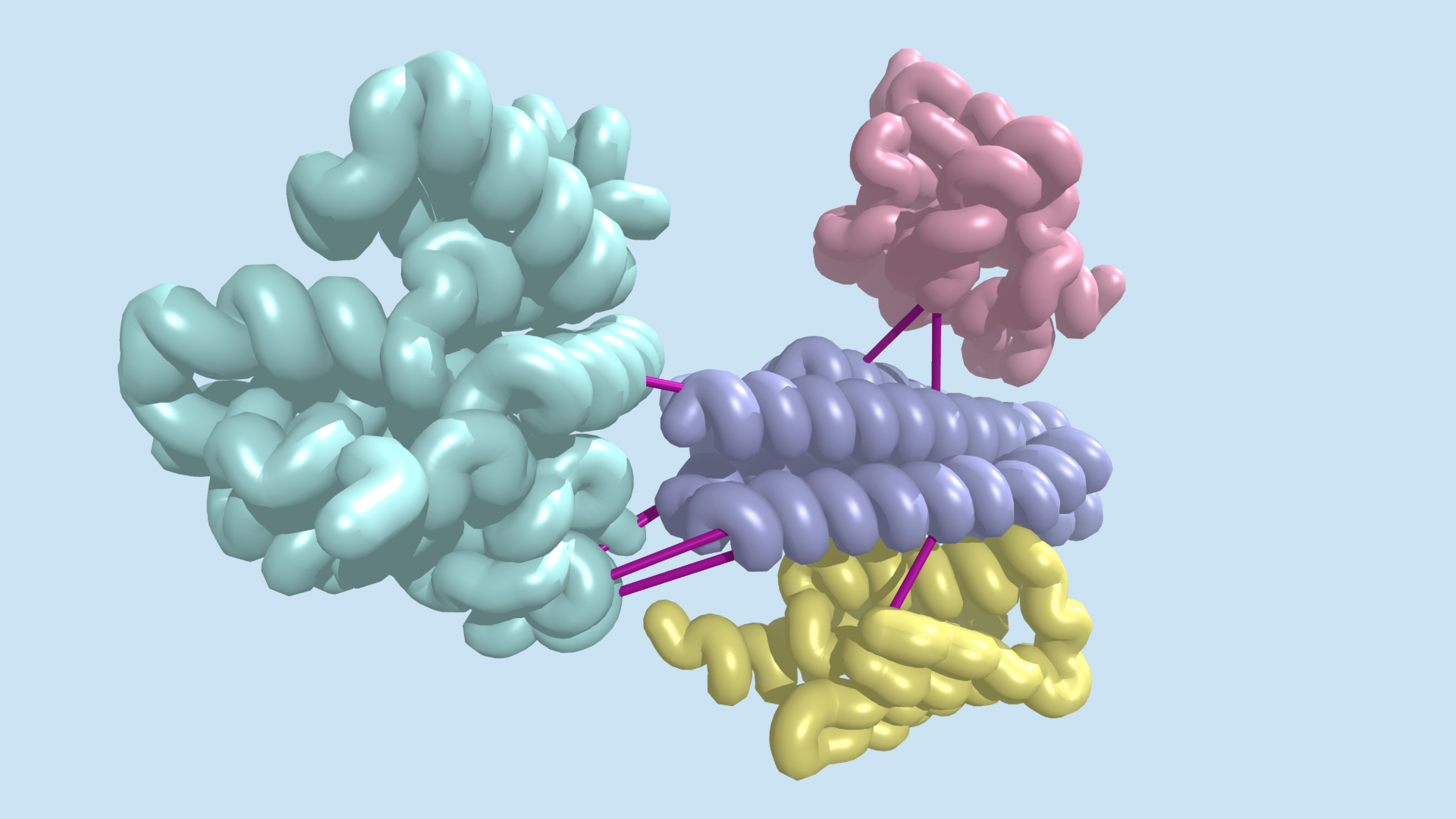

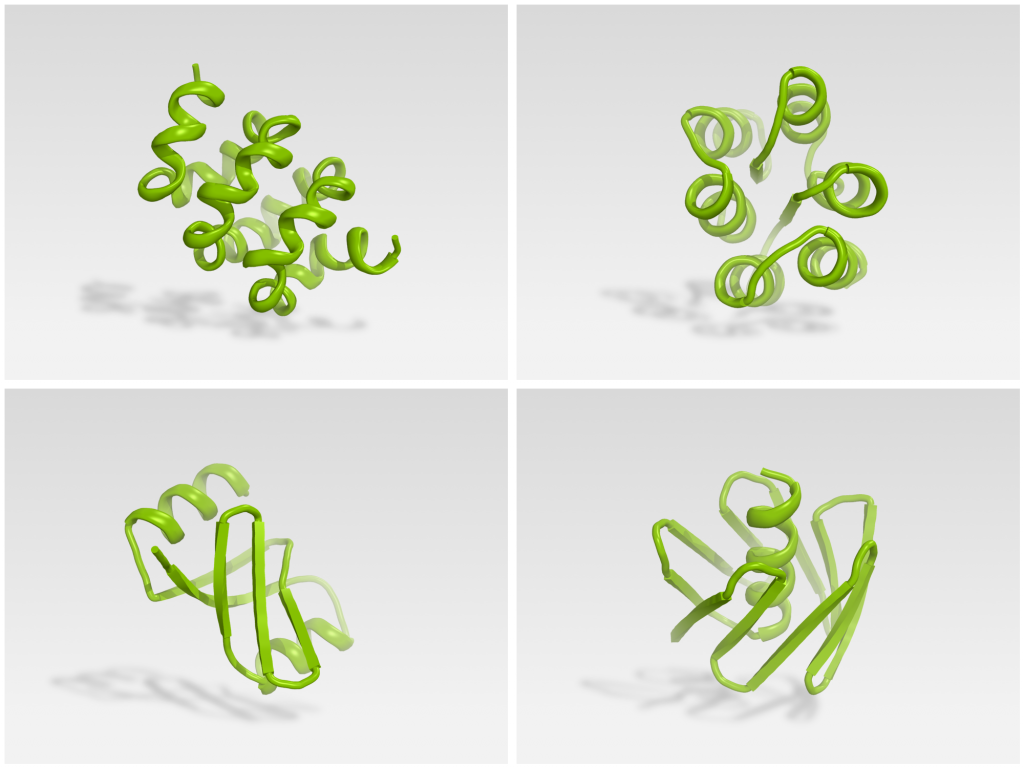
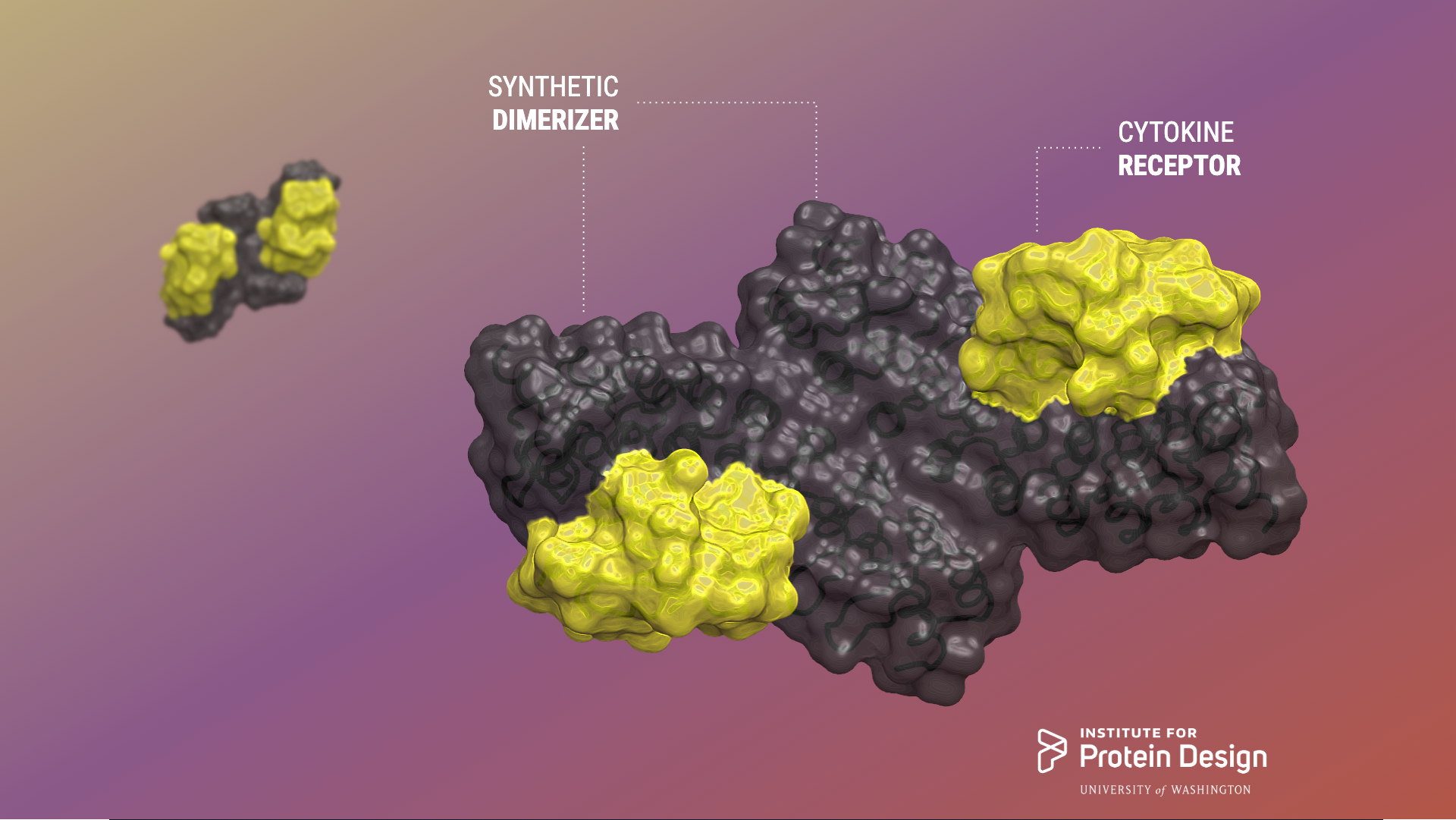
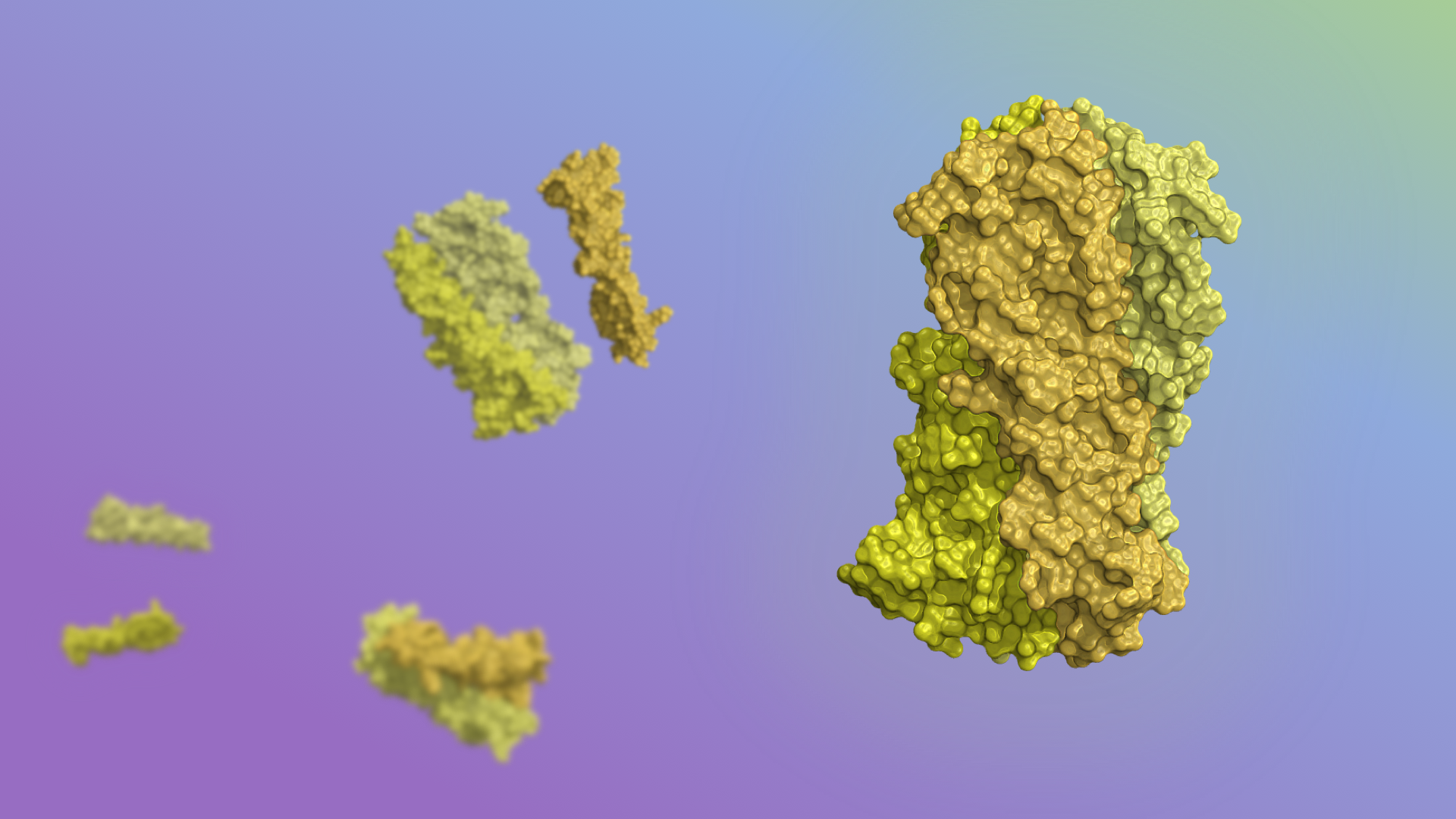
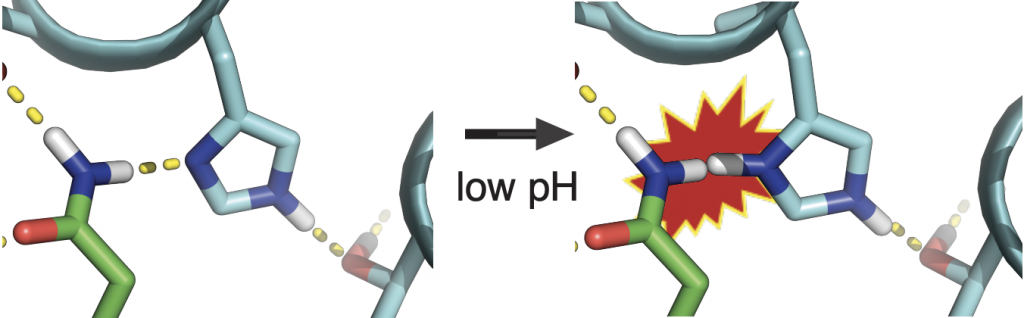

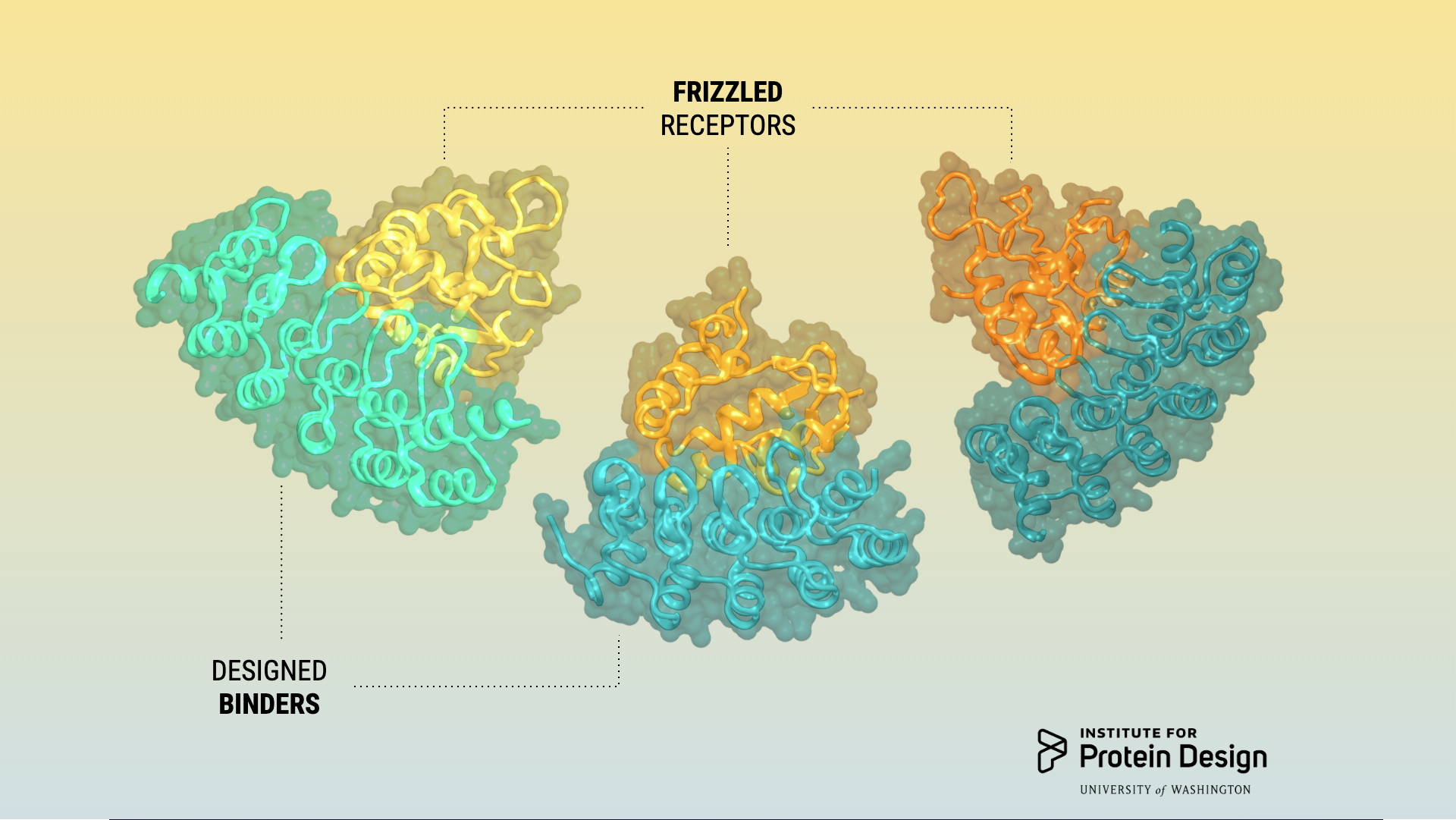



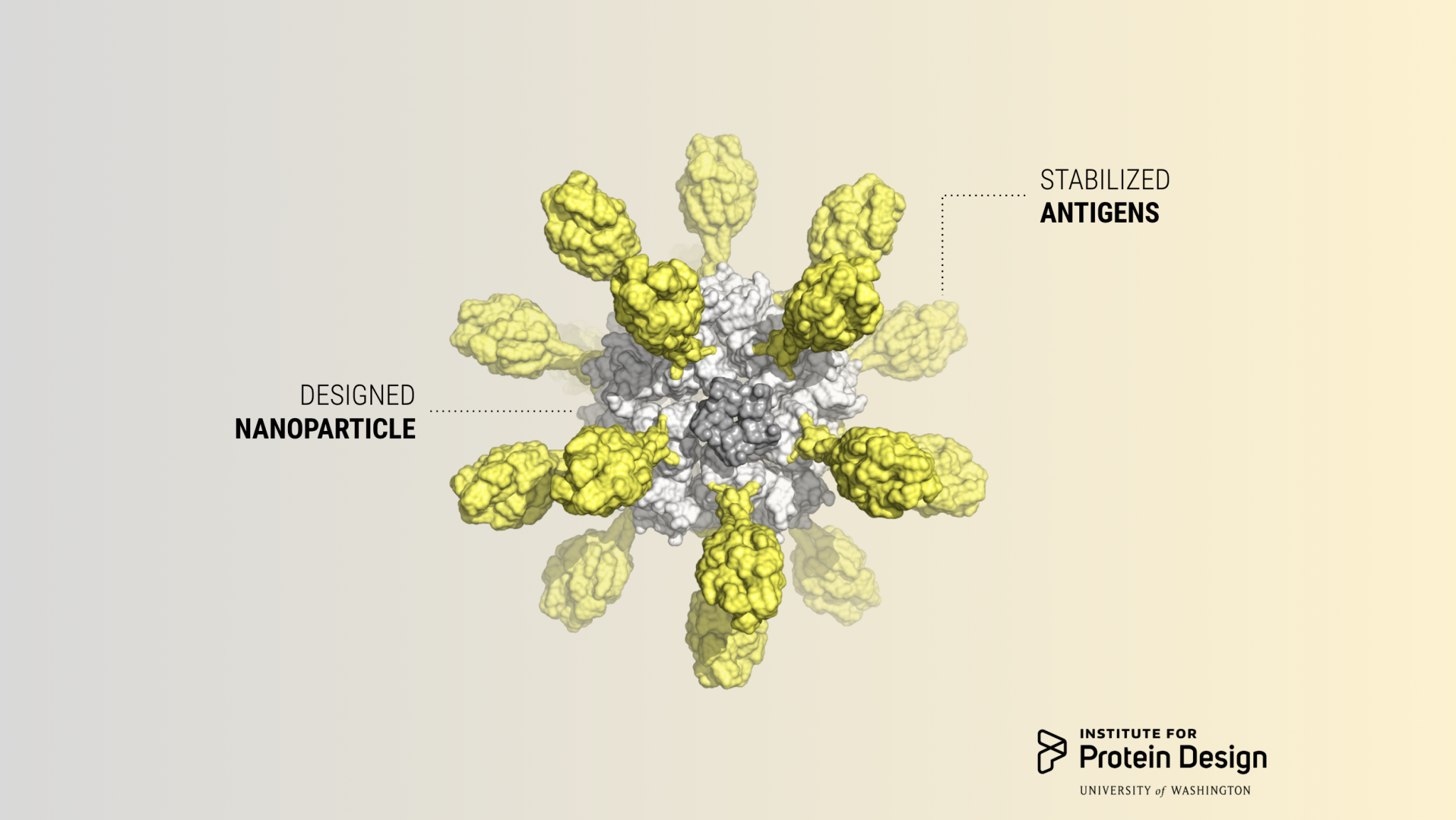

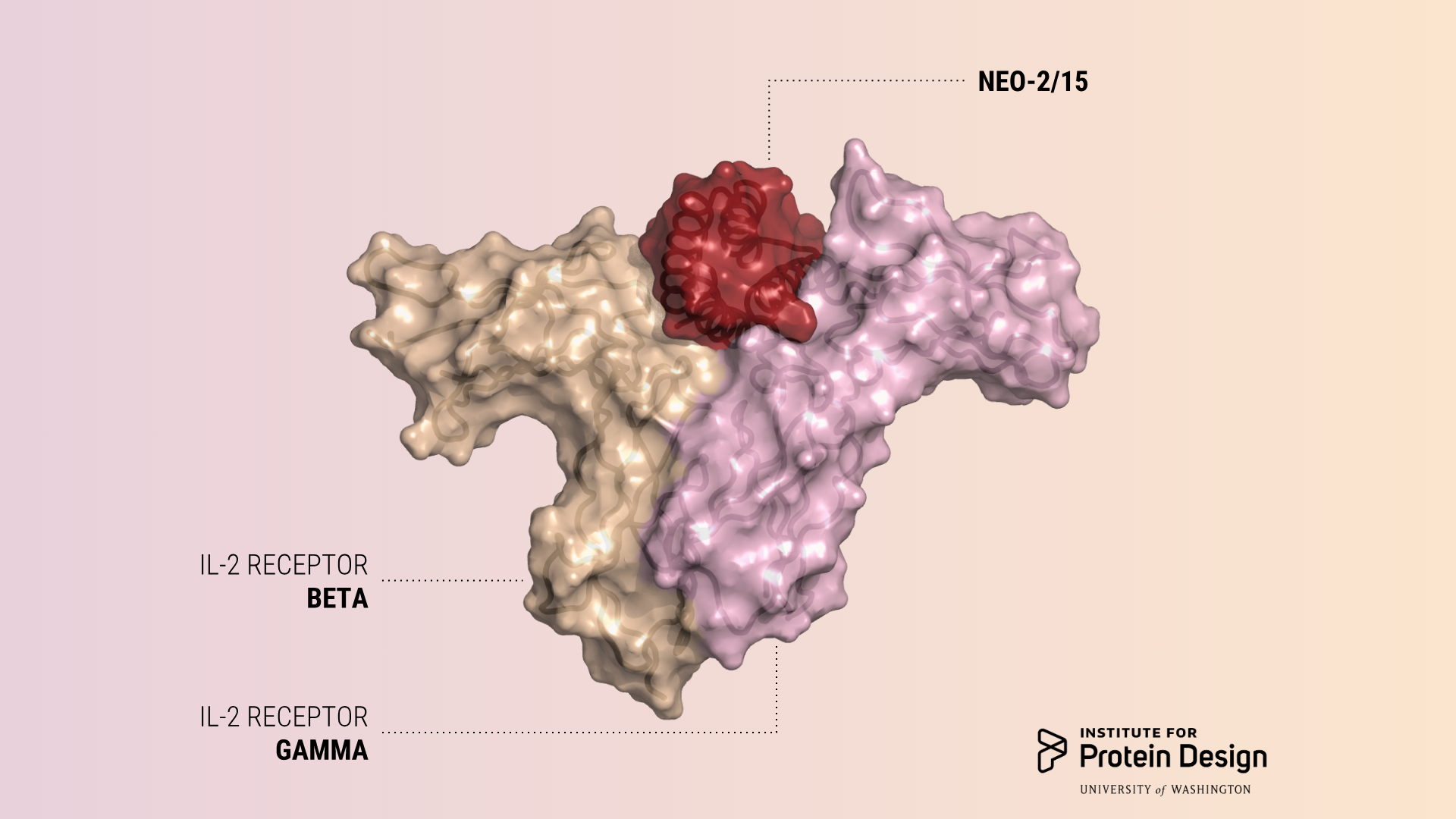
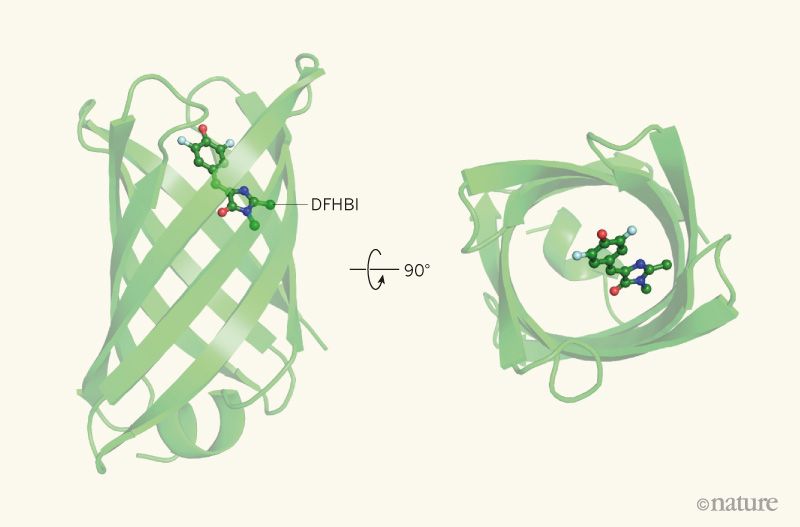
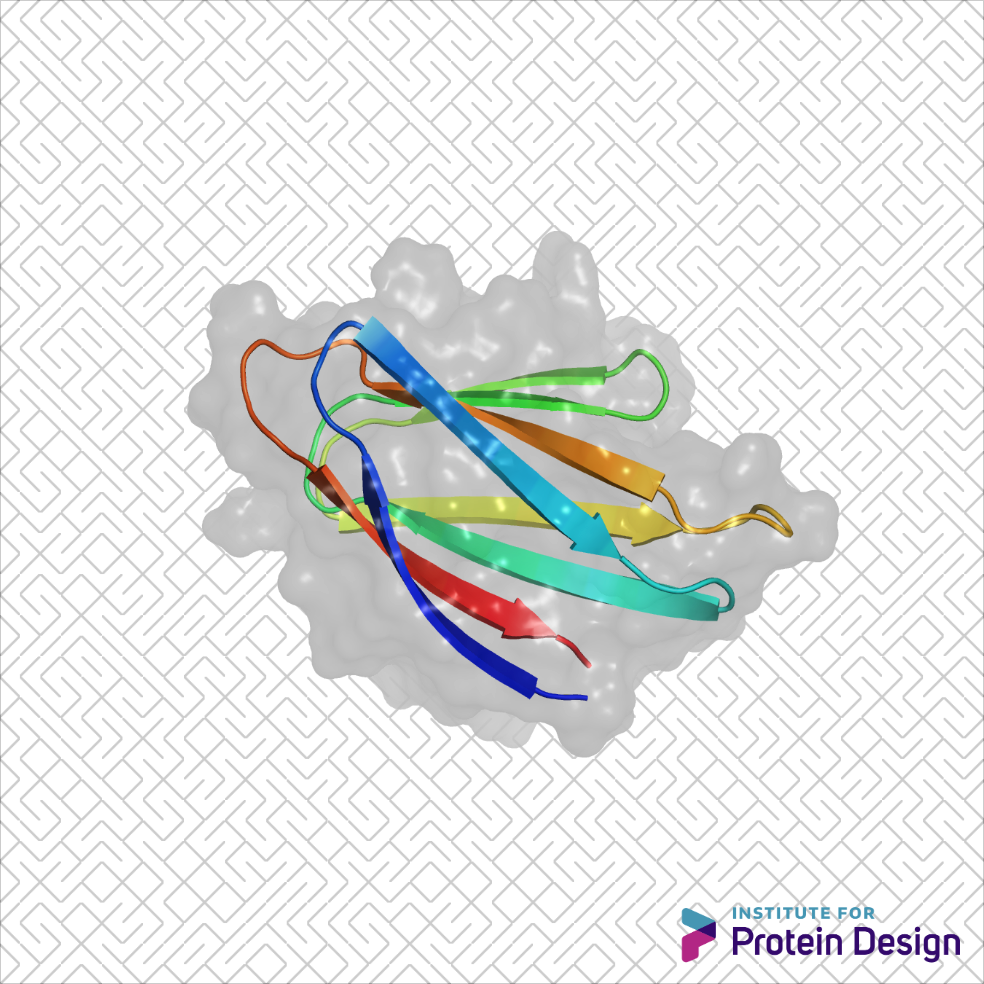


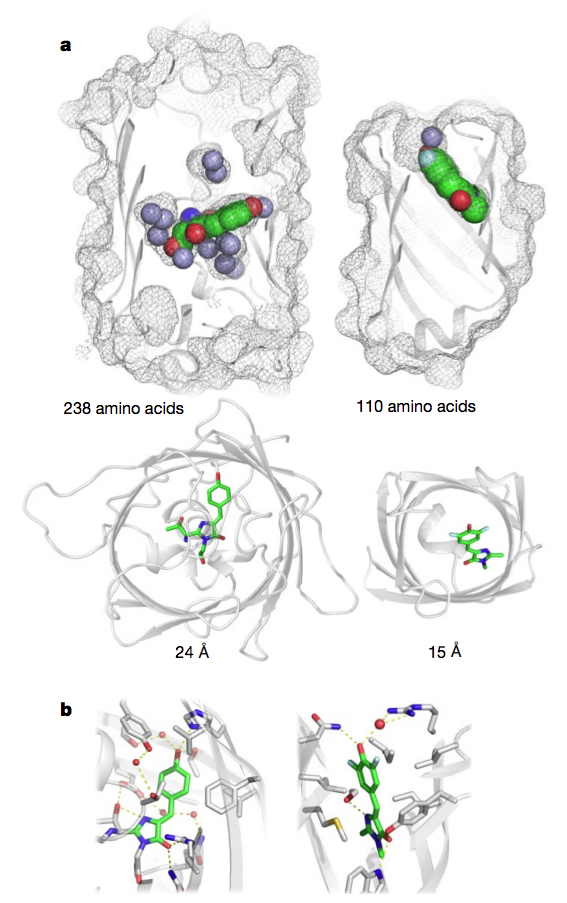




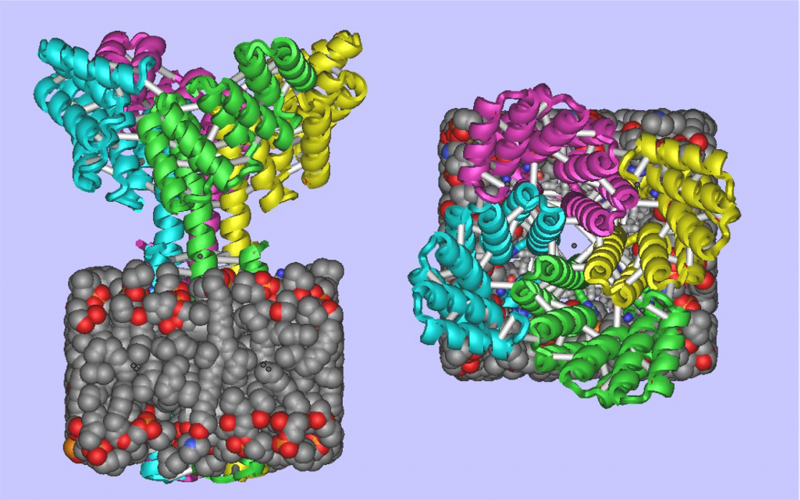
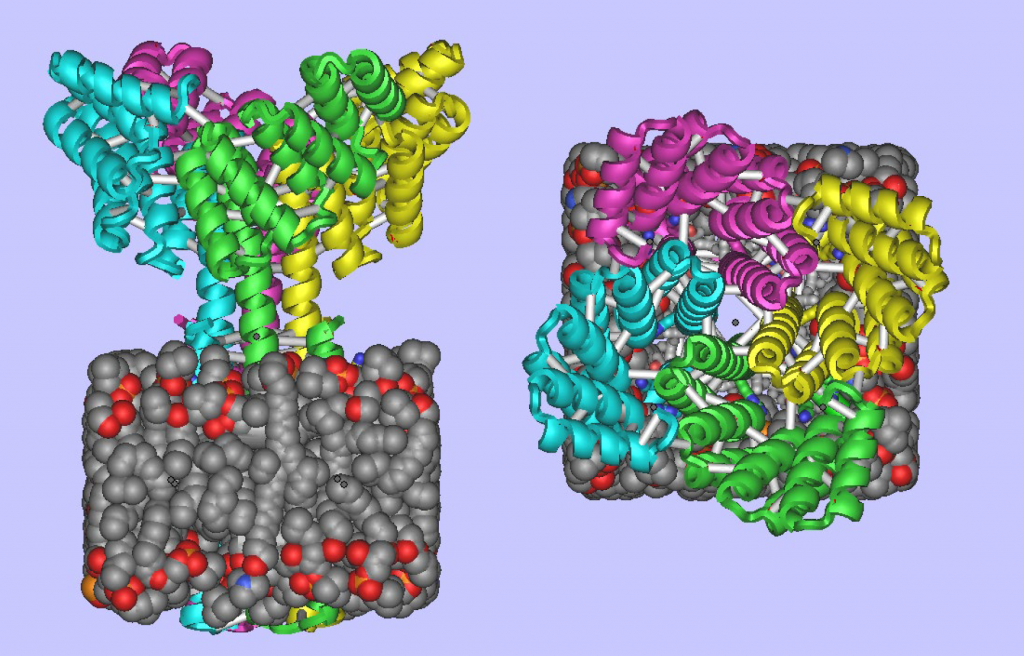
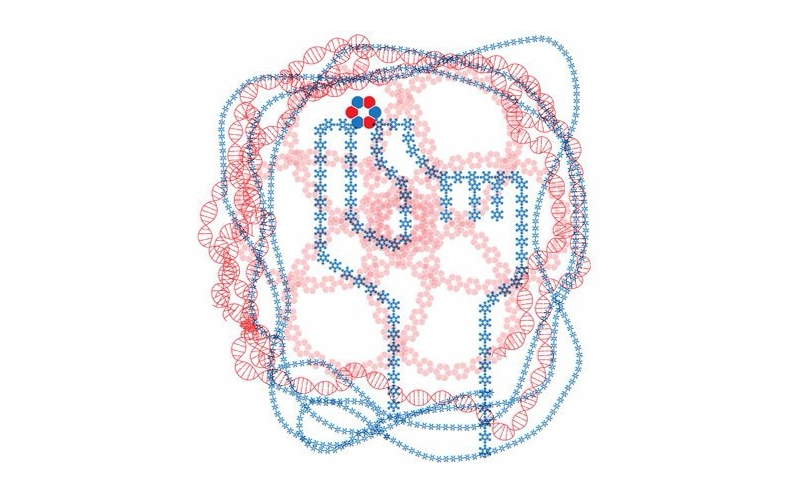
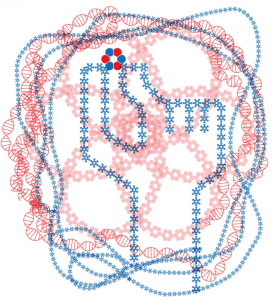
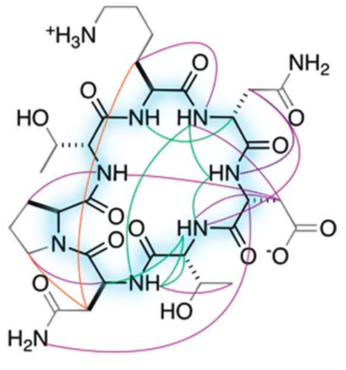
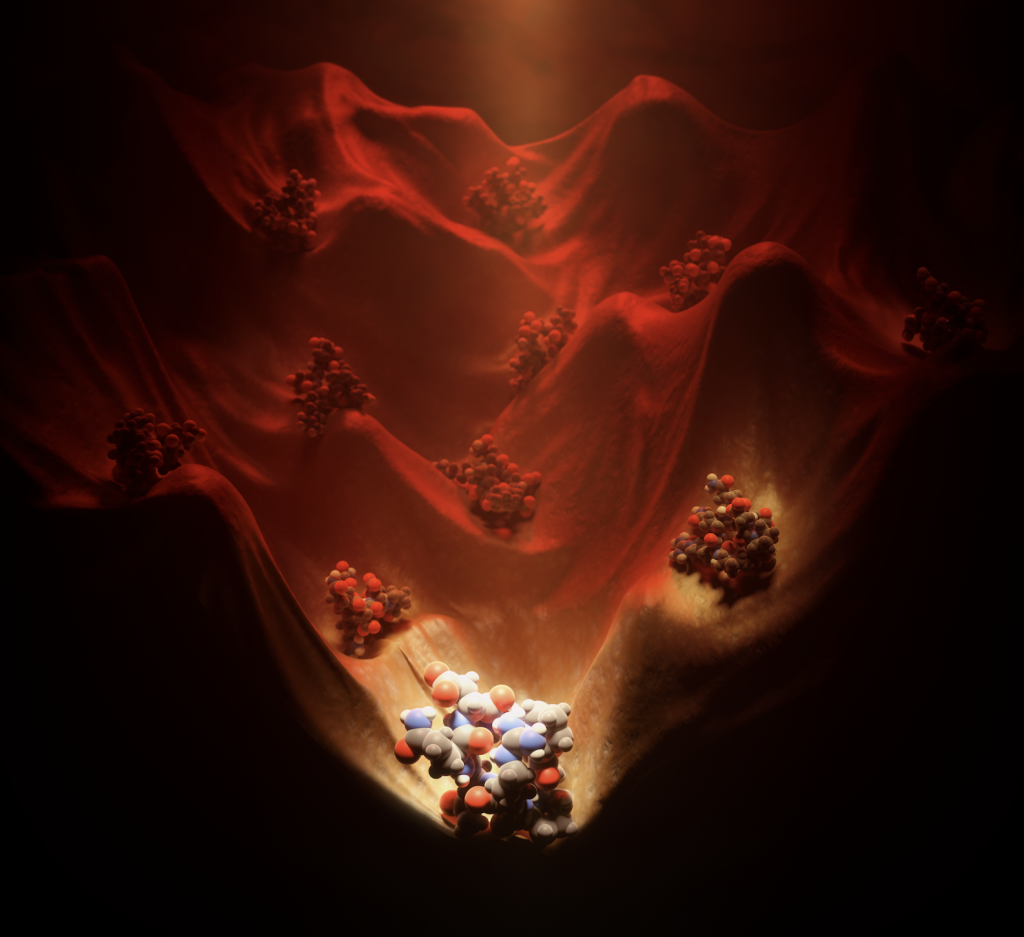
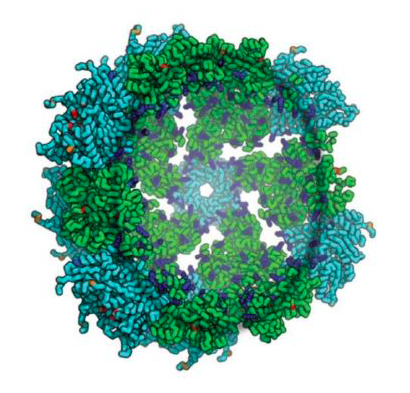








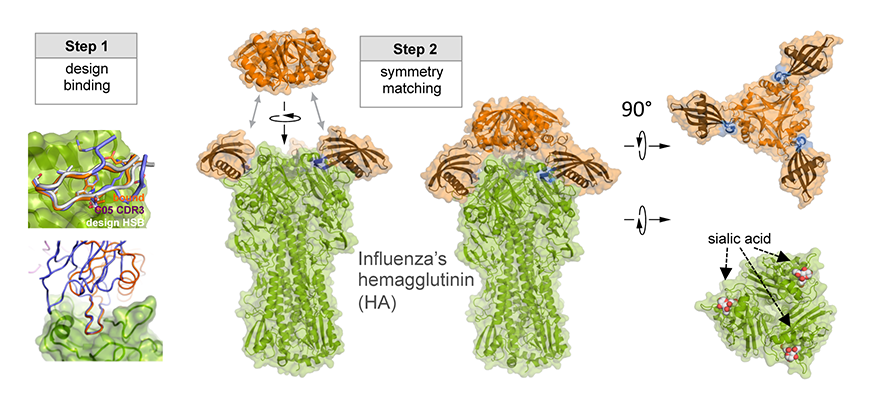
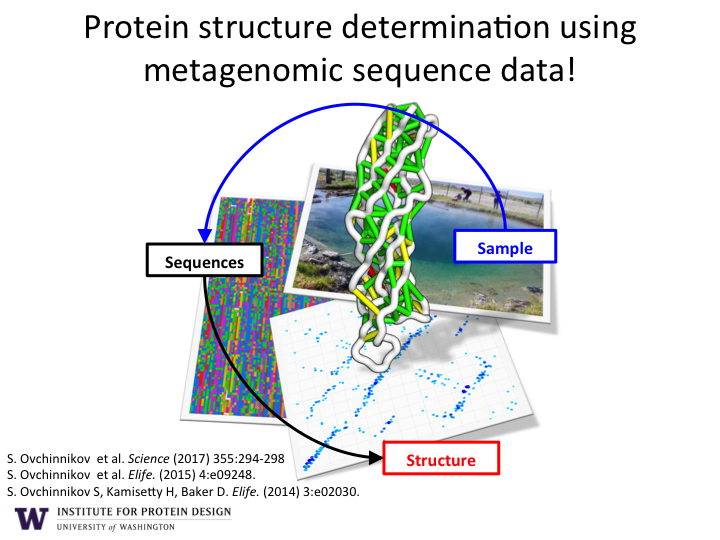
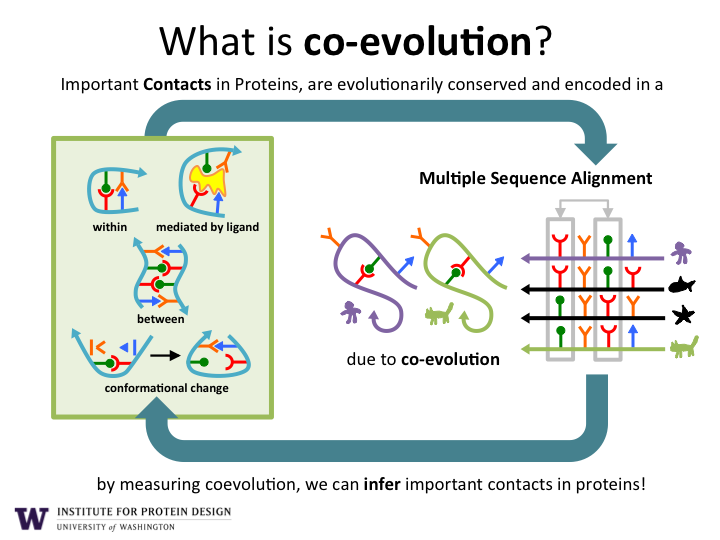
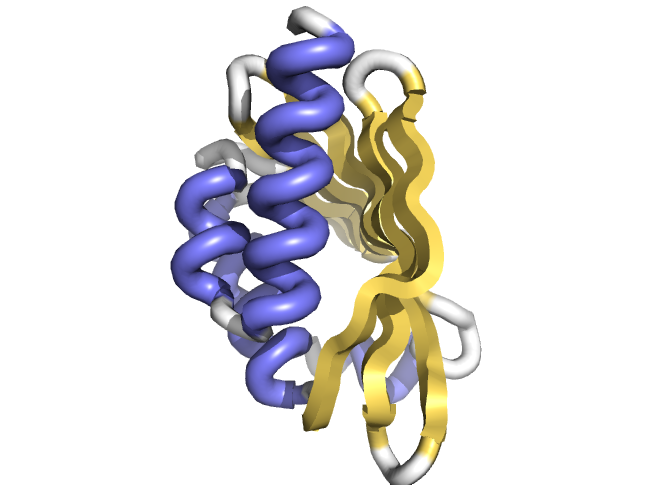




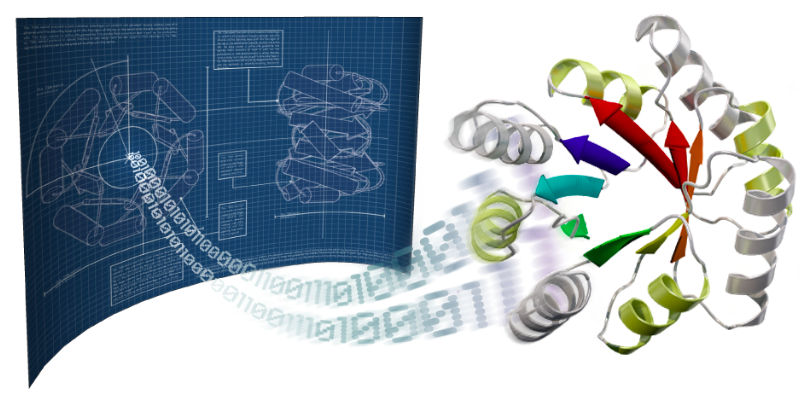
 Today is Limb Girdle Muscular Dystrophy Day, and the Institute for Protein Design is collaborating with the
Today is Limb Girdle Muscular Dystrophy Day, and the Institute for Protein Design is collaborating with the 


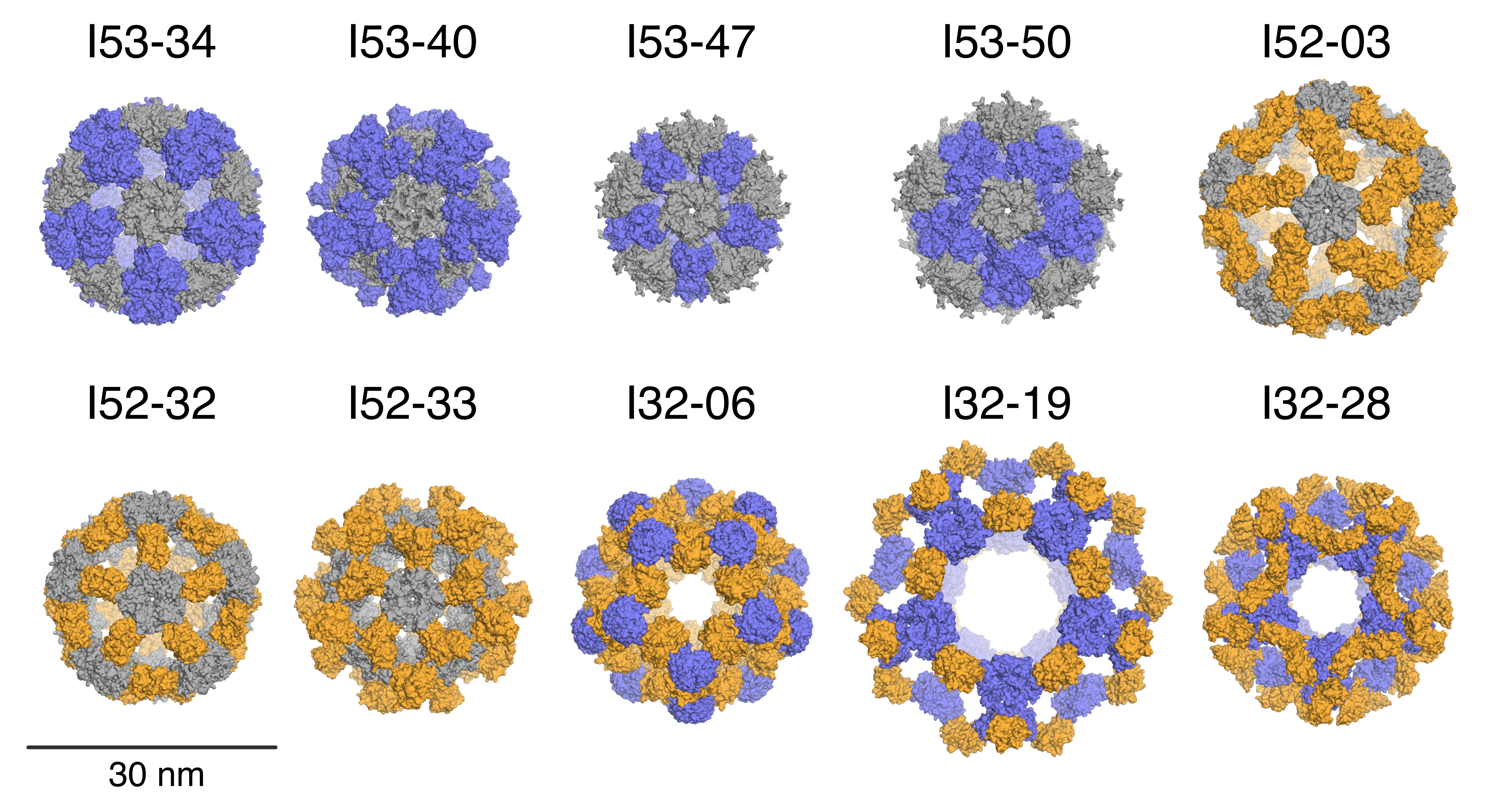

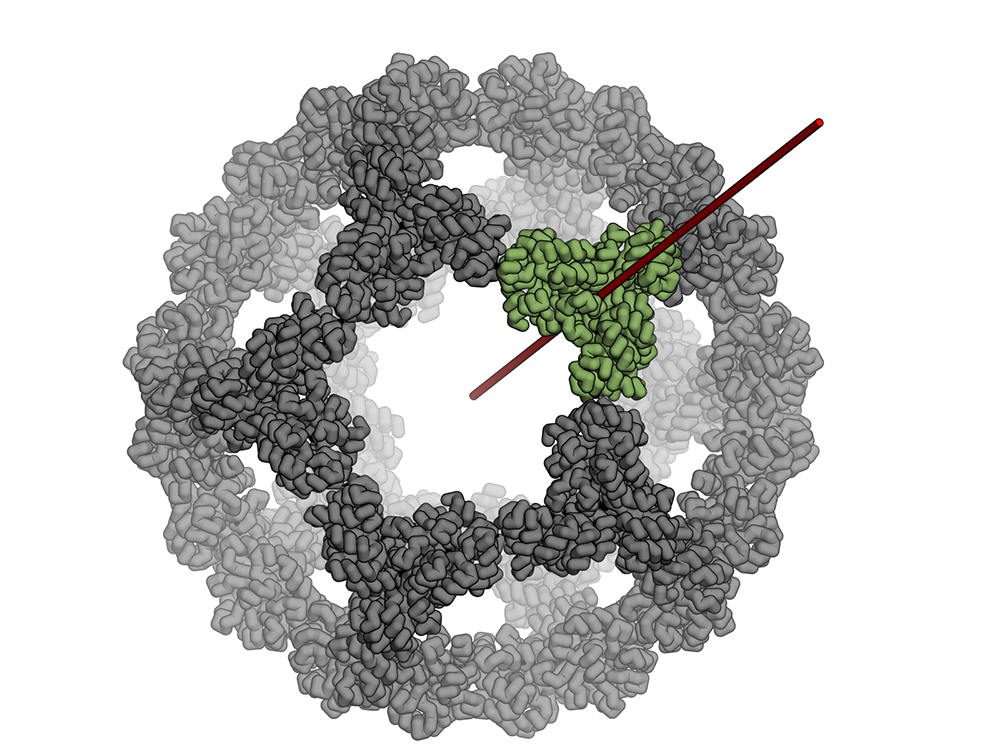

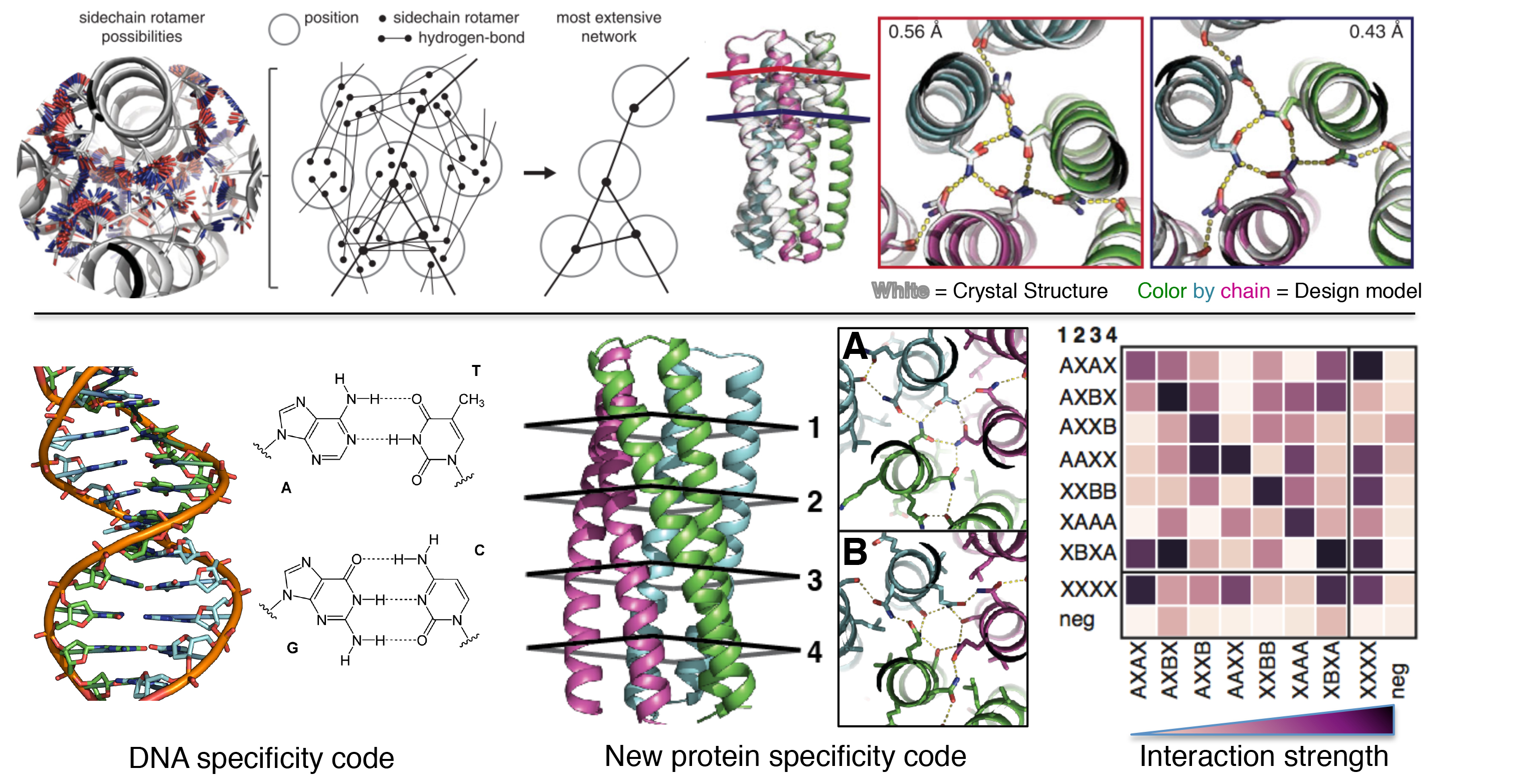





 As technology improves and Americans spend more on treatments to cure or prevent disease and injury, 2016 is likely to be a challenging year in health care. Doctors, nurses and clinicians are learning to work in new and innovative ways as consumers rely on video consults and their smartphones as diagnostic tools.
As technology improves and Americans spend more on treatments to cure or prevent disease and injury, 2016 is likely to be a challenging year in health care. Doctors, nurses and clinicians are learning to work in new and innovative ways as consumers rely on video consults and their smartphones as diagnostic tools.
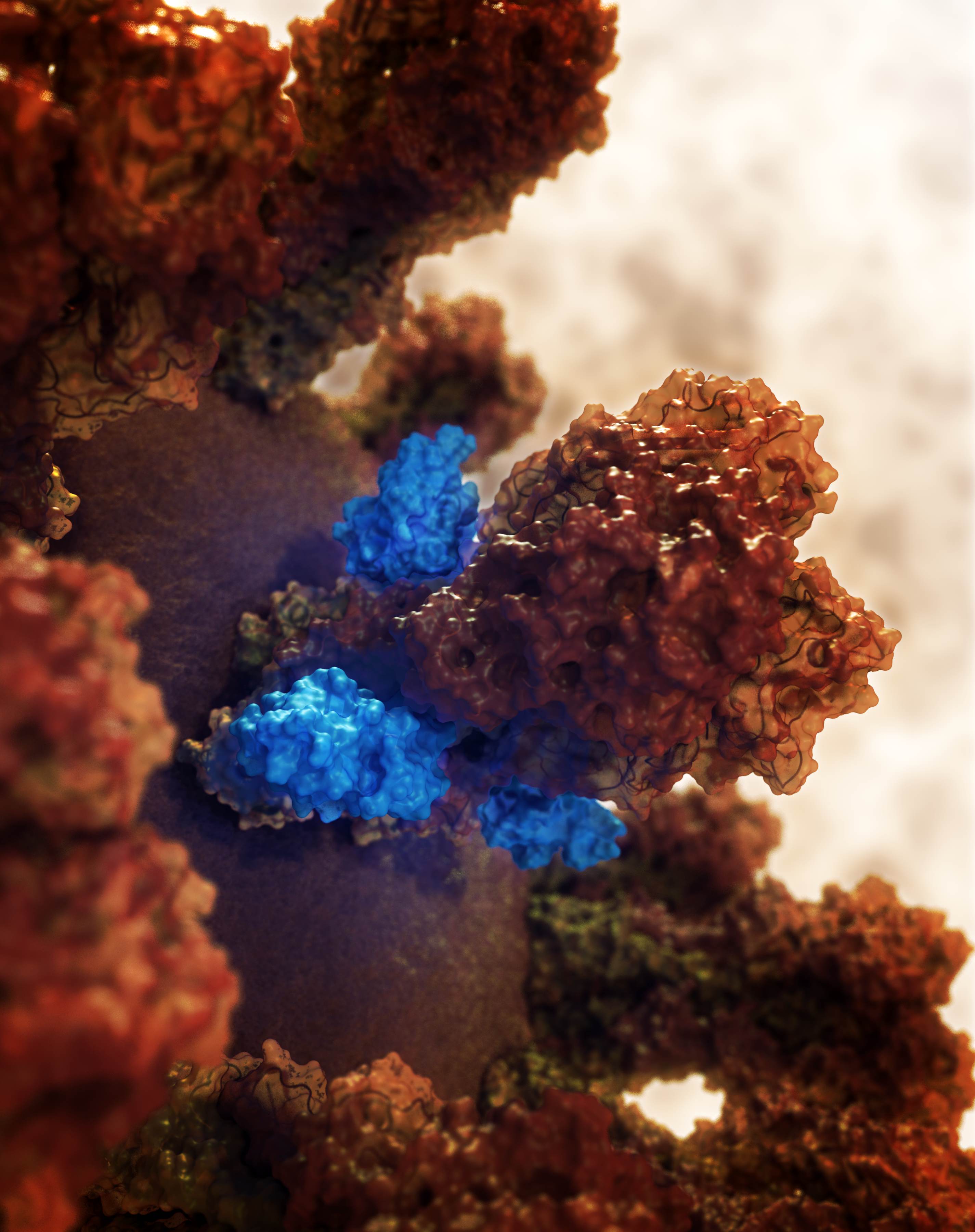

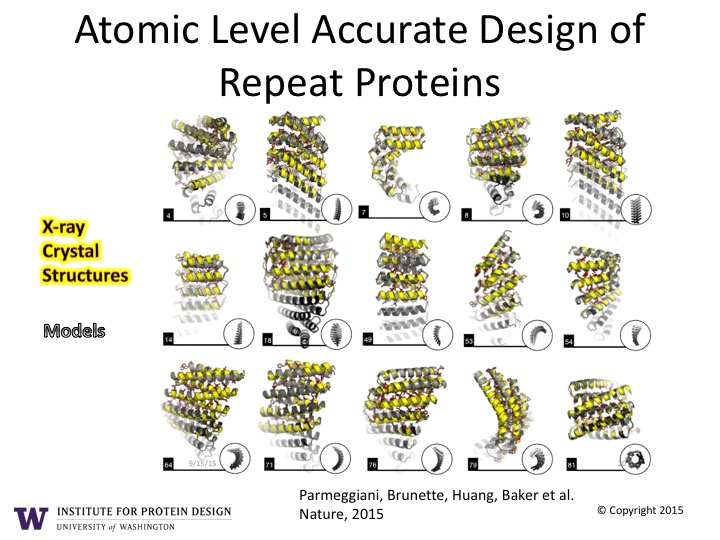
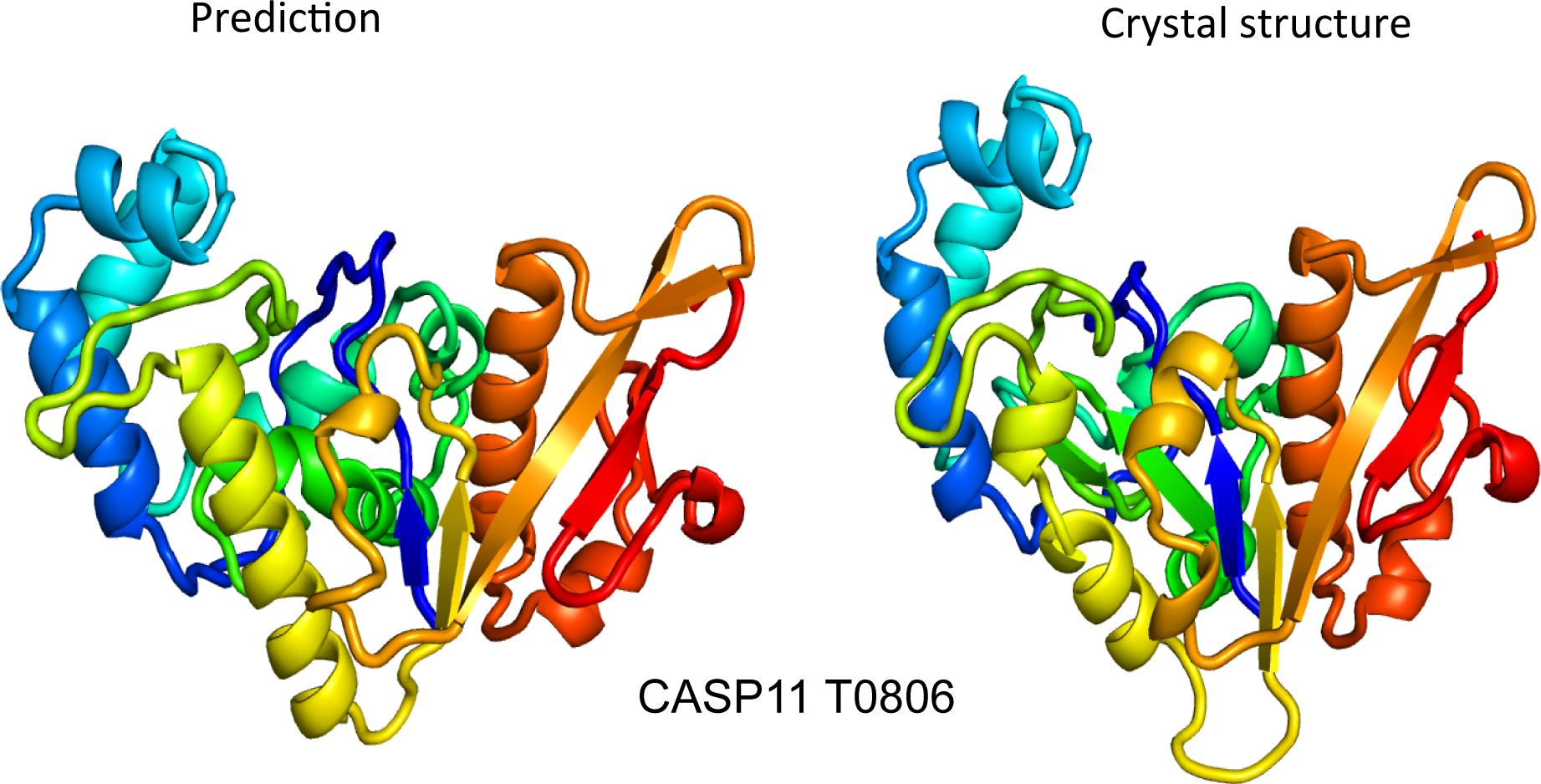
 In the early 1990s, researchers in the field of protein structure prediction were challenged by the problem of how to impartially judge the accuracy of prediction algorithms. This realization led the protein structure prediction the community to start the Critical Assessment of protein Structure Prediction (CASP), a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. In each CASP, an independent scientific advisory board solicits other researchers to submit experimentally verified, but unpublished, 3D protein structures to CASP. The linear amino acid sequences of these proteins are then provided to structure prediction researchers, who each have an equal and limited amount of time to submit final structure predictions to the CASP advisory board. The submitted structure predictions are then compared to the experimentally verified structures using the same metrics for all CASP contributors. Even though the primary goal of CASP is to help advance methods for identifying protein 3D structure given only its linear amino acid sequence, many view the experiment more as a “world championship” in protein structure prediction.
In the early 1990s, researchers in the field of protein structure prediction were challenged by the problem of how to impartially judge the accuracy of prediction algorithms. This realization led the protein structure prediction the community to start the Critical Assessment of protein Structure Prediction (CASP), a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. In each CASP, an independent scientific advisory board solicits other researchers to submit experimentally verified, but unpublished, 3D protein structures to CASP. The linear amino acid sequences of these proteins are then provided to structure prediction researchers, who each have an equal and limited amount of time to submit final structure predictions to the CASP advisory board. The submitted structure predictions are then compared to the experimentally verified structures using the same metrics for all CASP contributors. Even though the primary goal of CASP is to help advance methods for identifying protein 3D structure given only its linear amino acid sequence, many view the experiment more as a “world championship” in protein structure prediction. Using Rosetta to redesign the active site of the gliadin protease KumaMax – an enzyme computationally designed to break down gluten in the stomach – Dr. Pultz and collaborators show that the new variant Kuma030 degrades >99% of the gluten peptide that triggers inflammation in celiac disease patients. This work brings us even closer to arriving at an oral therapeutic for celiac disease.
Using Rosetta to redesign the active site of the gliadin protease KumaMax – an enzyme computationally designed to break down gluten in the stomach – Dr. Pultz and collaborators show that the new variant Kuma030 degrades >99% of the gluten peptide that triggers inflammation in celiac disease patients. This work brings us even closer to arriving at an oral therapeutic for celiac disease.


 INSTITUTE
INSTITUTE




 The exciting protein design work by IPD researchers and collaborators in PNAS titled
The exciting protein design work by IPD researchers and collaborators in PNAS titled 



 Happy Fall from the IPD! Read about our October happenings in this month’s news roundup. Click
Happy Fall from the IPD! Read about our October happenings in this month’s news roundup. Click  A new paper is out in this week’s issue of Science entitled High thermodynamic stability of parametrically designed helical bundles. Using novel computational design methods, extremely stable helical bundles can be custom designed with fine-tuned structural geometries for a number of applications. Read more about this exciting work at this
A new paper is out in this week’s issue of Science entitled High thermodynamic stability of parametrically designed helical bundles. Using novel computational design methods, extremely stable helical bundles can be custom designed with fine-tuned structural geometries for a number of applications. Read more about this exciting work at this 







